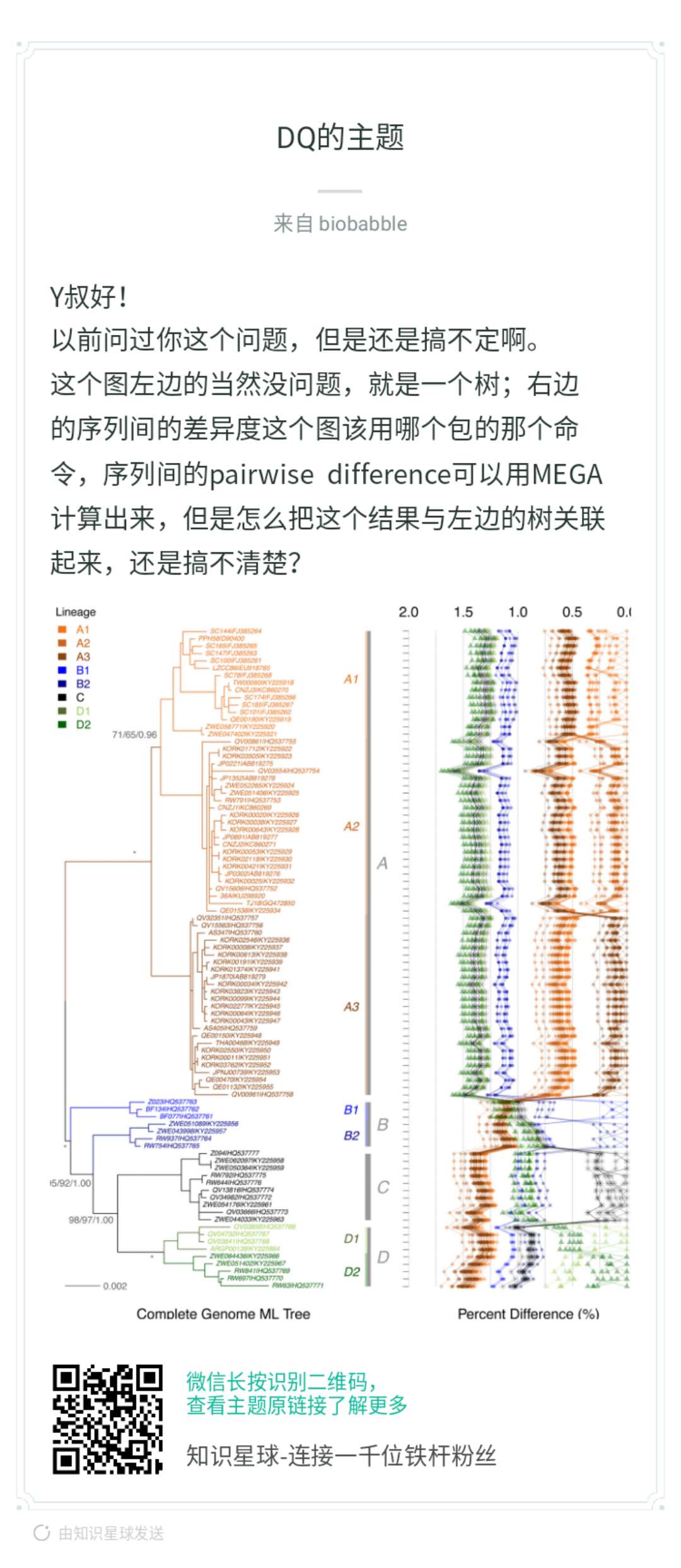
当然是ggplot2画,才好和ggtree兼容,你首先问自己,抛开树,你能右边吗?本身你有个矩阵,row的顺序要跟着树来,你可以先不管,先随意,这是y轴。那x轴呢?你矩阵里的数字,打点你总该会的,而点连线,同样column的数字打出来的点,就连在一起,你先按这个思路画出来,再来问后续的。
这是我在「知识星球」中的回答,然而小伙伴表示还是不懂。那么我来详细讲解一下:
首先我们读入一个FASTA数据:
require(treeio)
fasta <- system.file("examples/FluA_H3_AA.fas", package="ggtree")
aa <- read.fasta(fasta)
数据用treeio包里的read.fasta去读这个氨基酸序列,它有个好处是你直接as.character(aa[[i]])就是个字母的向量,方便比较。
n = length(aa)
d = matrix(NA, ncol=n, nrow=n)
nm = labels(aa)
rownames(d) = colnames(d) = nm
for (i in seq_len(n)) {
for (j in seq_len(i)) {
d[i, j] = mean(as.character(aa[[i]]) != as.character(aa[[j]]))
d[j, i] = d[i, j]
}
}
这段代码生成了pairwise distance矩阵,你可以搞其它的统计量,都OK的。大家可以想一想,一般这种矩阵怎么可视化?用热图!热图用数字来填充颜色,而现在我们把数字当成x轴上的变量,然后打点,仅此而已,我们有没有办法画出来,当然可以。同样row的数值会在同一y轴上,如果是热图,同一column的数值会在同一x轴上,方便横向和纵向比较,而现在同一column的数值不在同一直线一了,因为我们打的点的位置使用了矩阵中的数值,同一column的点被拉扯到不同的位置上去了,所以为了能够比较或者是看清楚同一column上的点的走势,我们把同一column上的点用线条连接在一起。这就是小伙伴提问的图,就是这么画出来的,非常简单。我在「知识星球」里其实已经解答得很清楚了。
Continue reading

