
multiple annotation in ChIPseeker
Nearest gene annotation
Almost all annotation software calculate the distance of a peak to the nearest TSS and assign the peak to that gene. This can be misleading, as binding sites might be located between two start sites of different genes or hit different genes which have the same TSS location in the genome.
The function annotatePeak provides option to assign genes with a max distance cutoff and all genes within this distance were reported for each peak.
> anno = annotatePeak(getSampleFiles()[[4]], addFlankGeneInfo=TRUE, flankDistance=5000)
>> loading peak file... 2014-09-30 01:12:09 PM
>> preparing features information... 2014-09-30 01:12:09 PM
>> identifying nearest features... 2014-09-30 01:12:10 PM
>> calculating distance from peak to TSS... 2014-09-30 01:12:11 PM
>> assigning genomic annotation... 2014-09-30 01:12:11 PM
>> adding flank feature information from peaks...2014-09-30 01:12:37 PM
>> assigning chromosome lengths 2014-09-30 01:12:39 PM
>> done... 2014-09-30 01:12:39 PM
> head(as.GRanges(anno))
GRanges object with 6 ranges and 14 metadata columns:
seqnames ranges strand | V4 V5
|
[1] chr1 [ 815092, 817883] * | MACS_peak_1 295.76
[2] chr1 [1243287, 1244338] * | MACS_peak_2 63.19
[3] chr1 [2979976, 2981228] * | MACS_peak_3 100.16
[4] chr1 [3566181, 3567876] * | MACS_peak_4 558.89
[5] chr1 [3816545, 3818111] * | MACS_peak_5 57.57
[6] chr1 [6304864, 6305704] * | MACS_peak_6 54.57
annotation geneChr geneStart geneEnd geneLength geneStrand
[1] Distal Intergenic chr1 803451 812182 8732 -
[2] Promoter (<=1kb) chr1 1243994 1247057 3064 +
[3] Promoter (<=1kb) chr1 2976181 2980350 4170 -
[4] Promoter (<=1kb) chr1 3547331 3566671 19341 -
[5] Promoter (<=1kb) chr1 3816968 3832011 15044 +
[6] Promoter (1-2kb) chr1 6304252 6305638 1387 +
geneId transcriptId distanceToTSS
[1] 284593 uc001abt.4 -5701
[2] 126789 uc001aed.3 0
[3] 440556 uc001aka.3 0
[4] 49856 uc001ako.3 0
[5] 100133612 uc001alg.3 0
[6] 390992 uc009vly.2 1452
flank_txIds
[1] uc001abt.4
[2] uc001aea.2;uc001aeb.2;uc001aec.1;uc001aed.3;uc010nyi.2;uc009vjx.3;uc009vjy.1;uc001aee.2;uc001aef.2;uc001aeg.2;uc001aeh.2;uc001aei.2;uc001aek.2;uc009vjz.2;uc010nyj.2
[3] uc001aka.3;uc010nzg.1;uc001akc.3;uc001ake.3;uc001akf.3;uc009vlh.3
[4] uc001ako.3;uc001akn.3;uc010nzi.2;uc001akp.3;uc021ofb.1;uc021ofc.1;uc021ofd.1;uc021ofe.1;uc021off.1
[5] uc001alg.3;uc010nzo.2;uc001alh.4;uc001alf.3;uc009vls.3;uc031pkz.1
[6] uc001amp.2;uc009vly.2
flank_geneIds
[1] 284593
[2] 116983;116983;116983;126789;126789;126789;54973;54973;54973;54973;54973;54973;54973;54973;54973
[3] 440556;440556;63976;63976;63976;63976
[4] 49856;49856;49856;7161;7161;7161;7161;7161;7161
[5] 100133612;100133612;100133612;339448;339448;339448
[6] 387509;390992
flank_gene_distances
[1] -2910
[2] -1978;-18;0;0;0;-128;8492;15729;15729;15729;15729;15729;15729;15729;15729
[3] 0;0;-4514;-4514;-4514;-4514
[4] 0;0;0;-1253;-1253;-1253;-1253;-1253;-1253
[5] 0;0;0;0;0;0
[6] 15331;0
-------
seqinfo: 24 sequences from hg19 genome
Three extra columns, flank_txIds, flank_geneIds, flank_gene_distances were added if parameter addFlankGeneInfo is set to TRUE.
Genomic annotation
The annotation column annotates the genomic location of a given peak. Since some annotations overlap, they are assigned based on the following priority.
- Promoter (defined by tssRegion parameter)
- 5' UTR
- 3' UTR
- Exon
- Intron
- Downstream (immediate downstream of a gene, within 3kb)
- Distal Intergenic
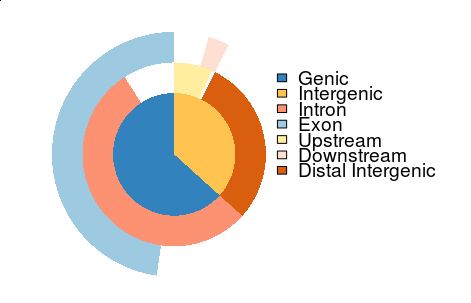
I have implemented plotAnnoPie and plotAnnoBar to visualize their distribution. I am wondering how to visualize the annotation overlap and found venn pie-agram can partially solve this issue. It’s hard/impossible to visualize all the overlap information within one vennpie plot.
I employed the vennpie to ChIPseeker, and now with the vennpie function, we can visualize some of the overlap annotations.
vennpie(anno)
In order to manage complex data, annotatePeak now output a S4 object. Users can use as.GRanges to convert it to as.GRanges object or as.data.frame to convert it to data.frame and then the output can be exported by write.table function.
Citation
Yu G, Wang LG and He QY*. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31(14):2382-2383.
