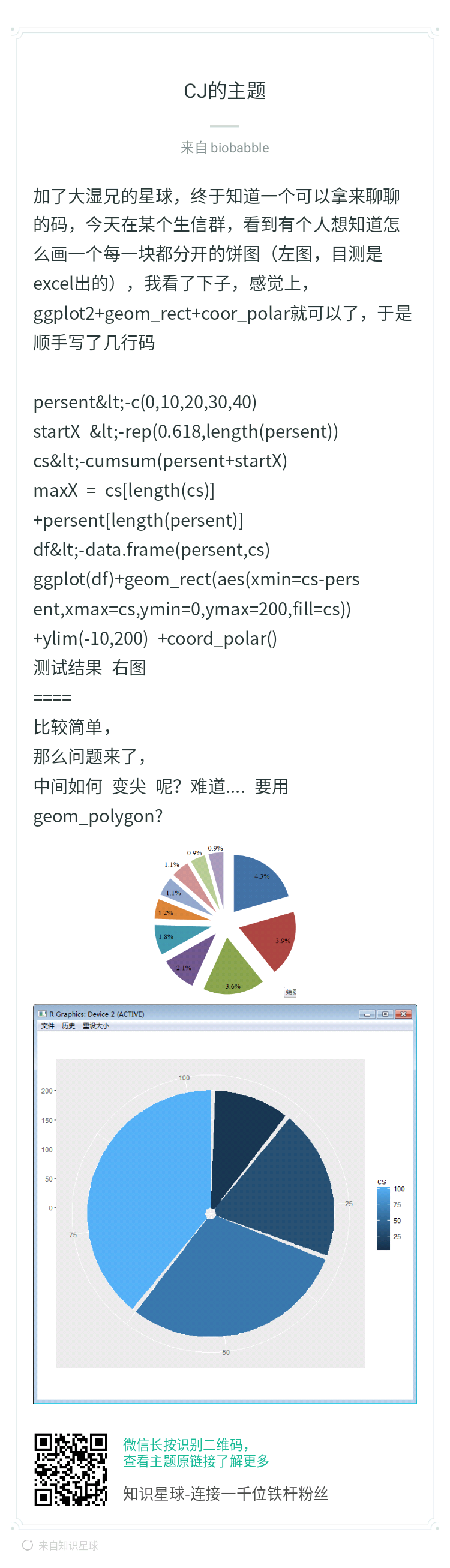
ggtree画树是不带root edge的,之前在《中空的环形树》一文中,有用户想要加长长的root edge,这样变成环形树的时候,就可以用这个root edge把树给顶出去,这个我给出的解决方案是留白,大家可以回过去看那篇文章。
然而root edge在某些时候还是需要的,我其实也一直有要写的想法,不过因为不是特别需要的东西,一直也就放着,刚好最近有用户在feature request,想要画root edge,于是我就实现了geom_rootedge图层,当你加这个图层的时候,root edge就自动画上去了。