
ggtree for microbiome data
ggtree can parse many software outputs and the evolution evidences inferred by these software can be used directly for tree annotation. ggtree not only works as an infrastructure that enables evolutionary data that inferred by commonly used software packages to be used in R, but also serves as a general tree visualization and annotation tool for the R community as it supports many S3/S4 objects defined by other R packages.
phyloseq for microbiome data
phyloseq class defined in the phyloseq package was designed for microbiome data. phyloseq package implemented plot_tree function using ggplot2. Although the function was implemented by ggplot2 and we can use theme, scale_color_manual etc for customization, the most valuable part of ggplot2, adding layer, is missing. plot_tree only provides limited parameters to control the output graph and it is hard to add layer unless user has expertise in both phyloseq and ggplot2.
library(phyloseq)
data(GlobalPatterns)
GP <- prune_taxa(taxa_sums(GlobalPatterns) > 0, GlobalPatterns)
GP.chl <- subset_taxa(GP, Phylum=="Chlamydiae")
plot_tree(GP.chl, color="SampleType", shape="Family", label.tips="Genus", size="Abundance") + ggtitle("tree annotation using phyloseq")
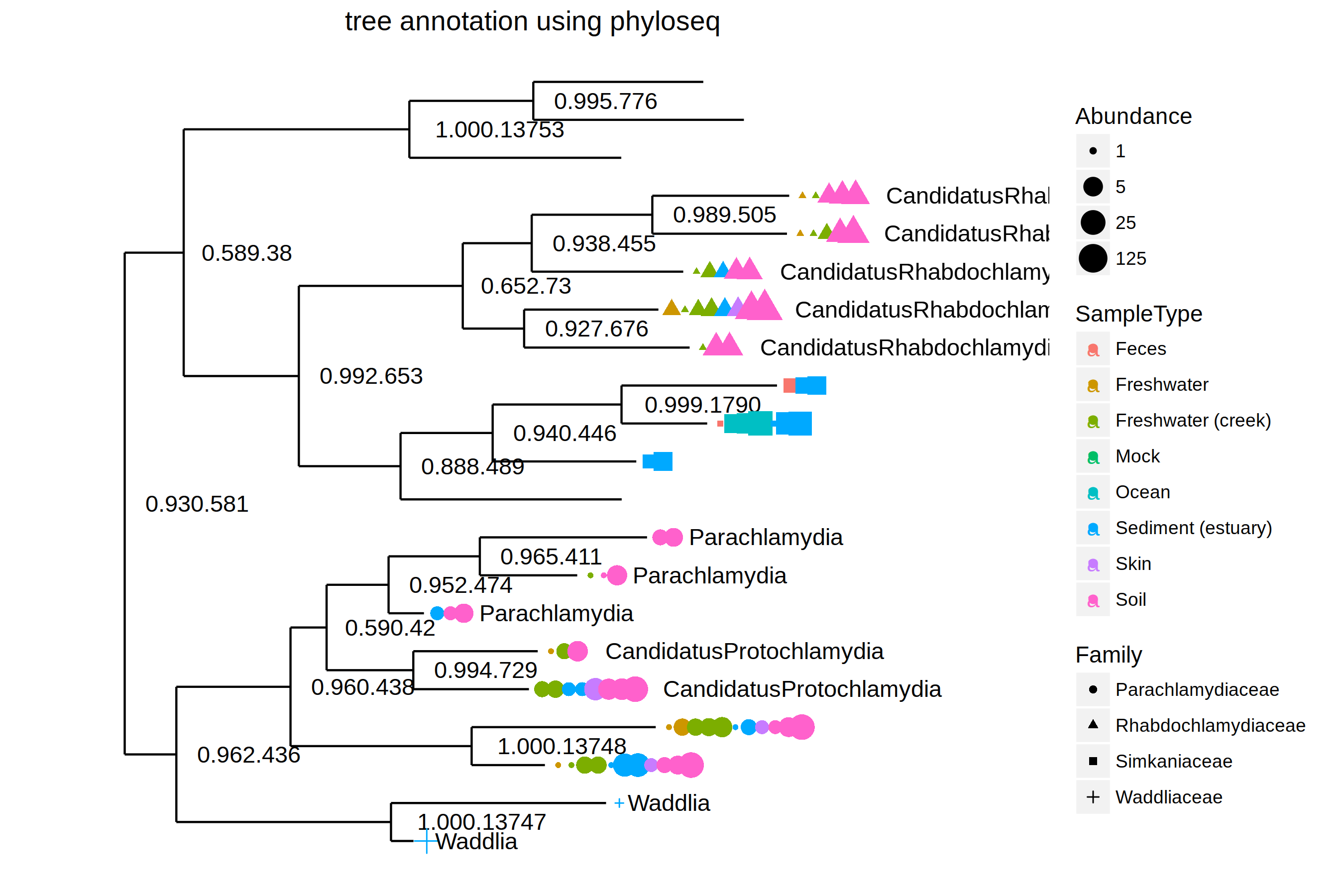
PS: If we look at the plot careful, we will find that legend produce by plot_tree is not correct (plot_tree map SampleType to color text which was shown in legend, but we can’t find the mapping in the plot).
ggtree supports phyloseq object
One of the advantage of R is the community. R users develop packages that can work together and complete each other. ggtree fits the R ecosystem in phylogenetic analysis. It supports several classes defined in other R packages that designed for storing phylogenetic tree with associated data, including phyloseq.
library(scales)
library(ggtree)
p <- ggtree(GP.chl, ladderize = FALSE) + geom_text2(aes(subset=!isTip, label=label), hjust=-.2, size=4) +
geom_tiplab(aes(label=Genus), hjust=-.3) +
geom_point(aes(x=x+hjust, color=SampleType, shape=Family, size=Abundance),na.rm=TRUE) +
scale_size_continuous(trans=log_trans(5)) +
theme(legend.position="right") + ggtitle("reproduce phyloseq by ggtree")
print(p)
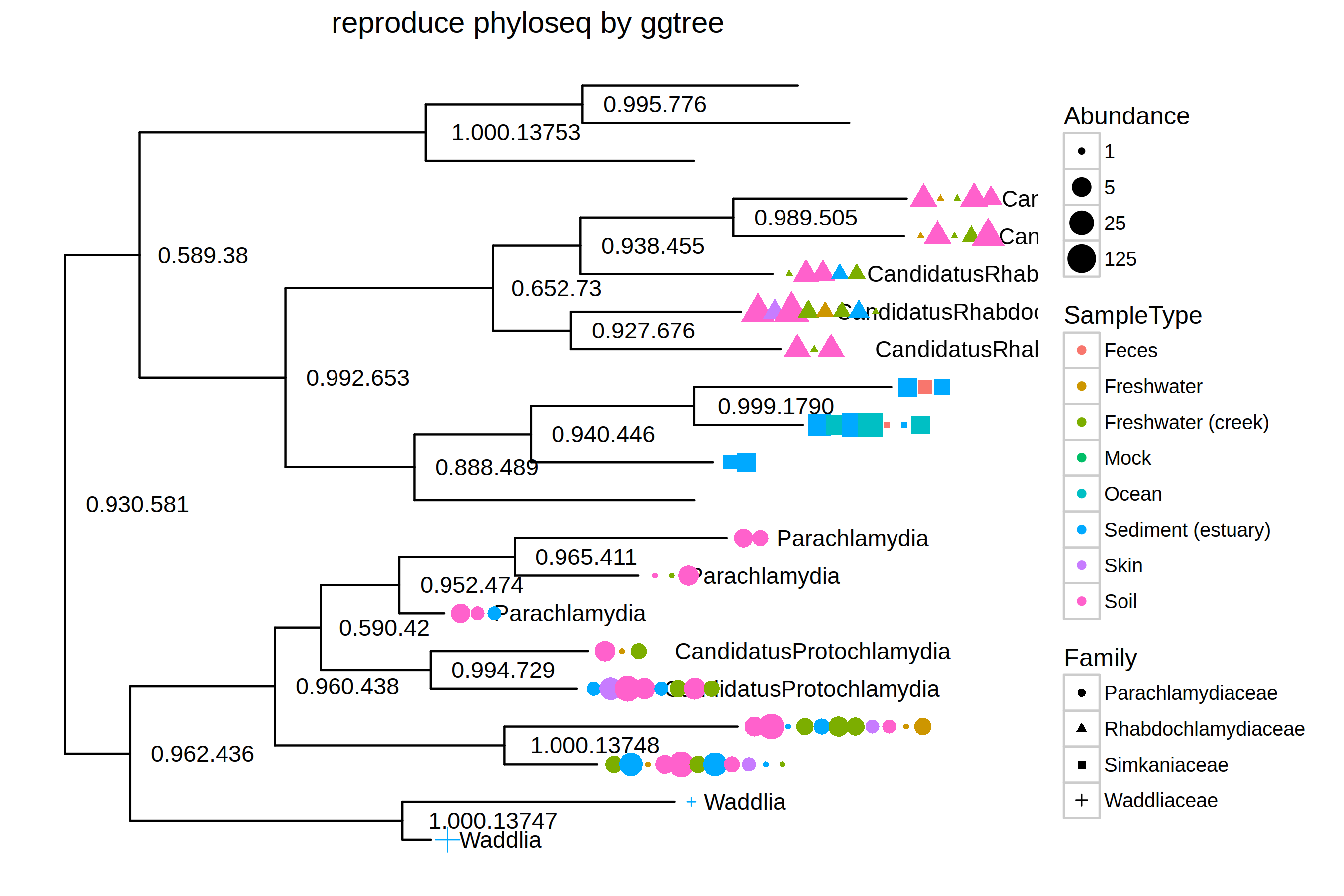
With ggtree, it would be more flexible to combine different layers using grammar of graphics syntax and more powerful since layers can be added without limitation (i.e. those predefined in plot_tree function). As an example, I extract the barcode sequence from the tree object and use msaplot to visualize the barcode sequence with the tree.
df <- fortify(GP.chl)
barcode <- as.character(df$Barcode_full_length)
names(barcode) <- df$label
barcode <- barcode[!is.na(barcode)]
msaplot(p, Biostrings::BStringSet(barcode), width=.3, offset=.05)
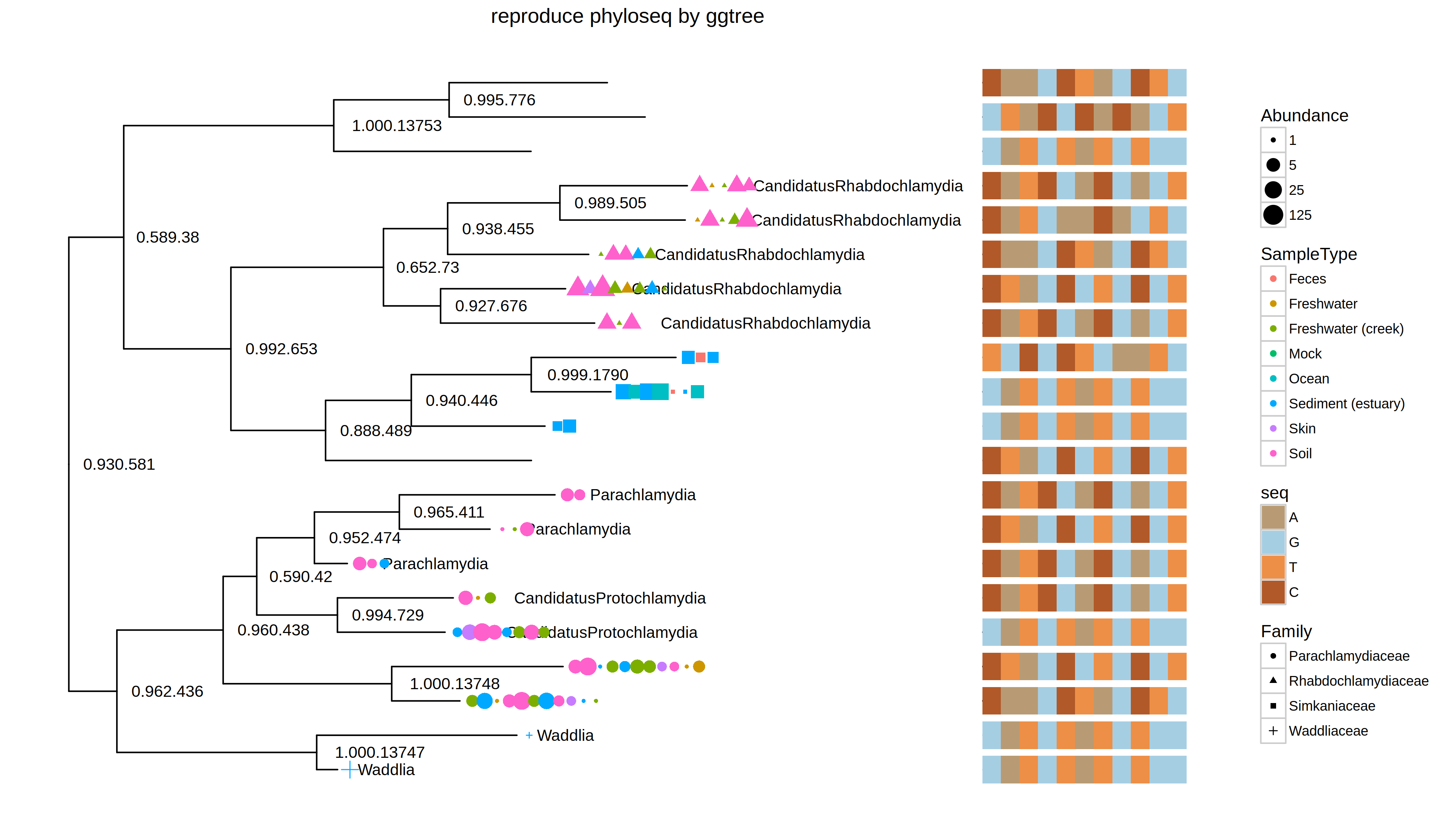
PS: I am thinking about writing a tutorial through examples. If you have any interesting topic, please let me know.
Citation
G Yu, DK Smith, H Zhu, Y Guan, TTY Lam*. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution. doi:10.1111/2041-210X.12628.
