
进化树和基因组结构
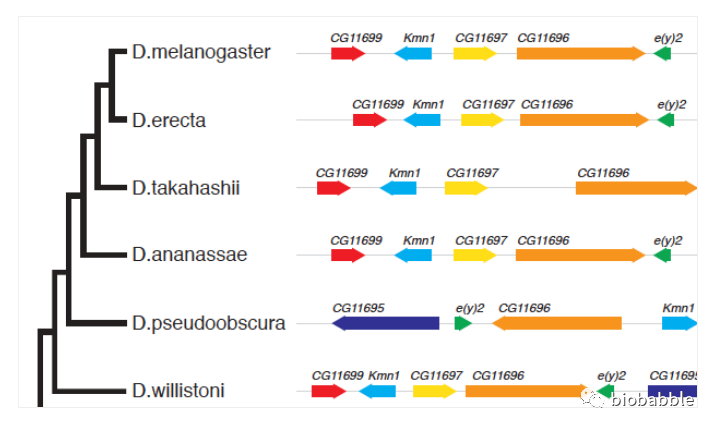
像上图这种高级货是可以使用ggtree来画的。
有一个叫gggenes的R包,画出来的图是这样子的。

但是这个包的图层不能直接使用《文章发表:两种方法在进化树上可视化数据》所提到的facet_plot(以及新版的geom_facet)来画。于是我在ggtree里写了一个图层叫geom_motif,包装一下,然后就可以愉快地使用了,而且让画基因组结构按照某个基因来对齐更加容易,只需要使用on参数,并且直接支持在上面label基因的名字。
废话少说,直接开撸上例子,这里使用gggenes自带的数据。当然这个数据没有配套的进化树,所以我们要无中生有创建一颗树出来。
library(dplyr)
library(ggplot2)
library(gggenes)
library(ggtree)
get_genes <- function(data, genome) {
filter(data, molecule == genome) %>% pull(gene)
}
g <- unique(example_genes[,1])
n <- length(g)
d <- matrix(nrow = n, ncol = n)
rownames(d) <- colnames(d) <- g
genes <- lapply(g, get_genes, data = example_genes)
上面定义一个函数,从数据中把归属于某个基因组的基因列表拿出来。然后应用于这个示例数据,拿到所有基因组的基因列表。我们通过这个列表来生成距离矩阵。
很简单,按照overlap了多少基因,算一下jaccard相似性,1-sim就得到dist。
for (i in 1:n) {
for (j in 1:i) {
jaccard_sim <- length(intersect(genes[[i]], genes[[j]])) /
length(union(genes[[i]], genes[[j]]))
d[j, i] <- d[i, j] <- 1 - jaccard_sim
}
}
有了距离矩阵,树也就可以生成了:
tree <- ape::bionj(d)
最后可视化也就跟着来了。
ggtree(tree, branch.length='none') +
geom_tiplab() + xlim_tree(5) +
geom_facet(mapping = aes(xmin = start, xmax = end, fill = gene),
data=example_genes, geom=geom_motif, panel='Alignment',
on = 'genE', label = 'gene') +
scale_fill_brewer(palette = "Set3")

