
关于ChIPseq注释的几个问题
为什么我要用某个基因组版本?
在上一篇文章中,我用了TxDb.Hsapiens.UCSC.hg19.knownGene。 hg19的TxDb, 或者有人就要问了,为什么不用hg38?
这个问题,不是说要用那一个,不能用那一个。而是你必须得用某一个,这取决于你最初fastq用BWA/Bowtie2比对于某个版本的基因组,你最初用了某个版本,后面就得用相应的版本,不能混,因为不同版本的位置信息有所不同。
当然如果要(贵圈喜欢的)强搞,也不是不可以,你得有chain file,先跑个liftOver,实际上就是在两个基因组版本之间做了位置转换。
为什么说ChIPseeker支持所有物种?
背景注释信息用了TxDb就能保证所有物种都支持了?我去哪里找我要的TxDb?
我写ChIPseeker的时候,我做的物种是人,ChIPseeker在线一周就有剑桥大学的人写信跟我说在用ChIPseeker做果蝇,在BED文件一文中,也提到了最近有人在Biostars上问用ChIPseeker做裂殖酵母。
首先Bioconductor提供了30个TxDb包,可以供我们使用,这当然只能覆盖到一小部分物种,我们的物种基因组信息,多半要从UCSC或者Ensembl获得,我敢说支持所有物种,就是因为UCSC和ensembl上所有的基因组都可以被ChIPseeker支持。
因为我们可以使用GenomicFeatures包函数来制作TxDb对象:
- makeTxDbFromUCSC: 通过UCSC在线制作TxDb
- makeTxDbFromBiomart: 通过ensembl在线制作TxDb
- makeTxDbFromGRanges:通过GRanges对象制作TxDb
- makeTxDbFromGFF:通过解析GFF文件制作TxDb
比如我想用人的参考基因信息来做注释,我们可以直接在线从UCSC生成TxDb:
require(GenomicFeatures)
hg19.refseq.db <- makeTxDbFromUCSC(genome="hg19", table="refGene")
比如最近在biostars上有用户问到的,做裂殖酵母的注释,我们可以下载相应的GFF文件,然后通过makeTxDbFromGFF函数生成TxDb对象,像下面的命令所演示,spombe就是生成的TxDb,就可以拿来做裂殖酵母的ChIPseq注释。
download.file("ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosaccharomyces_pombe.chr.gff3", "schizosaccharomyces_pombe.chr.gff3")
require(GenomicFeatures)
spombe <- makeTxDbFromGFF("schizosaccharomyces_pombe.chr.gff3")
所以我敢说,所有物种都支持。像Johns Hopkins出品的CisGenome就只支持到12个物种而已,极大地限制了它的应用。
ChIPseeker有什么不能注释的吗?
这个我还没想到,像CpG是不支持的,但也有人「黑」出来了:
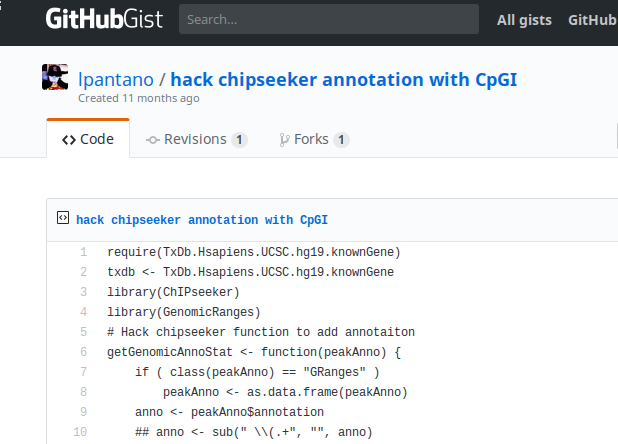
当然他的做法是把CpG也整合进去,如果你单纯只想看那些peak落在CpG上,或者说离CpG最近,不需要「黑」也能做到的,因为annotatePeak的背景注释信息除了TxDb之外,其实它还可以是自定义的GRanges对象,这保证了用户各种各样的需求,因为TxDb也不是万能的,如果能自定义,比如说我就只想看蛋白的结合位点会不会在内含子和外显子的交界处,再比如说我做的并不是编码蛋白的基因表达调控,而是非编码RNA,那么我想要用lncRNA的位置信息来做注释。像这样的需求,ChIPseeker都是可以满足的。
可以按正负链分开注释吗?
上一篇文章中就有人问了,能否同时给出正负链上最近的基因。首先ChIPseq数据通常情况下是没有正负链信息的(有特殊的实验可以有),annotatePeak函数有参数是sameStrand,默认是FALSE,你可以给你的peak分别赋正负链,然后传入sameStrand=TRUE,分开做两次,你就可以分开拿到正链和负链的最近基因。
最近基因位置是相对于TSS的,能否相对于整个基因?
答案也是可以的!
首先如果peak和TSS有overlap,genomic annotation就是promoter,而最近基因也是同一个,所以在这种情况下,两种注释都指向同一个基因,可以说信息是冗余的,能不能不要冗余信息?这个是可以的,你可以传入参数ignoreOverlap=TRUE,那么最近基因就会去找不overlap的。
最近基因是相对于TSS,如果和TSS有overlap,距离是0, 必须是最近。回到标题的问题,如果我想说只要和基因有overlap就是最近基因,这种情况其实你的最近基因就是host gene,也就是annotation这个column给出来的是相对应的,我们就想找peak所在位置的基因信息,那么这当然也是可以的,默认参数overlap=“TSS”, 如果改为overlap=“all”,它看的就是整个基因而不是TSS,当然distanceToTSS也还是会计算,如果overlap的不是TSS,而是基因体里,并不会因为而设为0。
如果我只想注释上游或者是下游的基因呢?
当然也可以,我们有ignoreUpstream和ignoreDownstream参数,默认都是FALSE。随便你想看上游还是下游,都可以。
为什么要有这么多参数?
我在前一篇,只讲了输入,输出,你知道两个输入,会看输出,你就可以做ChIPseq注释了,非常简单。但是我不能把annotatePeak能做的全列出来,会让大家觉得复杂。(而且简单的情况是最常见的行为)
在大家知道输入输出,觉得简单之后,再讲一讲,它有一些参数,可以应对别的情况,这些情况可能并不是我们做ChIPseq所需要的,但不同参数的灵活组合,是可以解决和应对不同的需求的。
比如说, DNA是可以断的,如果我们要对(DNA breakpoints)断点位置做注释,优先是overlap基因,再者是上游,你会发现很多软件都不能做了,但ChIPseeker可以。
做软件关键是要注重细节,不单注重自己需求的细节,更主要是要注重别人的细节


