
peak注释
这一次讲解非常重要的peak注释,注释在ChIPseeker里只需要用到一个函数annotatePeak,它可以满足大家各方面的需求。
输入
当然需要我们上次讲到的BED文件,ChIPseeker自带了5个BED文件,用getSampleFiles()可以拿到文件的全路径,它返回的是个named list,我这里取第4个文件来演示。annotatePeak的输入也可以是GRanges对象,你如果用R做peak calling的话,直接就可以衔接上ChIPseeker了。
> require(ChIPseeker)
> f = getSampleFiles()[[4]]
巧妇难为无米之炊,就像拿到fastq要跑BWA,你需要全基因组的序列一样,做注释当然需要注释信息,基因的起始终止,基因有那些内含子,外显子,以及它们的起始终止,非编码区的位置,功能元件的位置等各种信息。
很多软件会针对特定的物种去整理这些信息供软件使用,但这样就限制了软件的物种支持,有些开发者写软件本意也是解决自己的问题,可能对自己的研究无关的物种也没兴趣去支持。
然而ChIPseeker支持所有的物种,你没有看错,ChIPseeker没有物种限制,当然这是有前提的,物种本身起码是有基因的位置这些注释信息,不然就变无米之炊了。
这里我们需要的是一个TxDb对象,这个TxDb就包含了我们需要的各种信息,ChIPseeker会把信息抽取出来,用于注释时使用。
> require(TxDb.Hsapiens.UCSC.hg19.knownGene)
> txdb = TxDb.Hsapiens.UCSC.hg19.knownGene
> x = annotatePeak(f, tssRegion=c(-1000, 1000), TxDb=txdb)
>> loading peak file... 2017-03-09 11:29:18 PM
>> preparing features information... 2017-03-09 11:29:18 PM
>> identifying nearest features... 2017-03-09 11:29:19 PM
>> calculating distance from peak to TSS... 2017-03-09 11:29:20 PM
>> assigning genomic annotation... 2017-03-09 11:29:20 PM
>> assigning chromosome lengths 2017-03-09 11:29:42 PM
>> done... 2017-03-09 11:29:42 PM
这里需要注意的是,启动子区域是没有明确的定义的,所以你可能需要指定tssRegion,把基因起始转录位点的上下游区域来做为启动子区域。
有了这两个输入(BED文件和TxDb对象),你就可以跑注释了,然后就可以出结果了。
输出
如果在R里打输出的对象,它会告诉我们ChIPseq的位点落在基因组上什么样的区域,分布情况如何。
> x
Annotated peaks generated by ChIPseeker
1331/1331 peaks were annotated
Genomic Annotation Summary:
Feature Frequency
9 Promoter 48.1592787
4 5' UTR 0.7513148
3 3' UTR 4.2073629
1 1st Exon 0.7513148
7 Other Exon 3.9068370
2 1st Intron 3.6814425
8 Other Intron 7.7385424
6 Downstream (<=3kb) 1.1269722
5 Distal Intergenic 29.6769346
如果我想看具体的信息呢?你可以用as.GRanges方法,这里我只打印前三行:
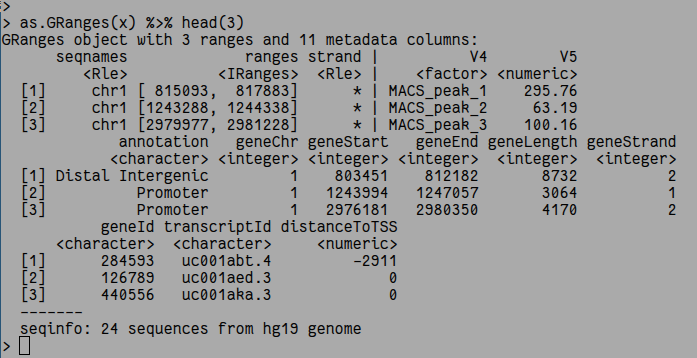
Bioconductor里有很多包是针对GRanges对象的,这样方便你在R里做后续的处理,如果你说你不懂这些,只想输出个Excel表格。那么也很容易,用as.data.frame就可以转成data.frame,然后你就可以用write.table输出表格了。
两种不同的注释
这里注释有两种,一种是genomic annotation (也就是annotation这一列)还有就是nearest gene annotation(也就是多出来的其它列)。
经常有人问我问题,把这两种搞混。genomic annotation注释的是peak的位置,它落在什么地方了,可以是UTR,可以是内含子或者外显子。
而最近基因是peak相对于转录起始位点的距离,不管这个peak是落在内含子或者别的什么位置上,即使它落在基因间区上,我都能够找到一个离它最近的基因(即使它可能非常远)。
这两种注释的策略是不一样的。针对不同的问题。第一种策略peak所在位置,可能就是调控的根本,比如你要做可变剪切的,最近基因的注释显然不是你关注的点。
而做基因表达调控的,当然promoter区域是重点,离结合位点最近的基因更有可能被调控。
最近基因的注释信息虽然是以基因为单位给出,但我们针对的是转录起始位点来计算距离,针对于不同的转录本,一个基因可能有多个转录起始位点,所以注释是在转录本的水平上进行的,我们可以看到输出有一列是transcriptId.
第三种注释
两种注释有时候还不够,我想看peak上下游某个范围内(比如说-5k到5k的距离)都有什么基因,annotatePeak也可以做到。
你只要传个参数说你要这个信息,还有什么区间内,就可以了。
x = annotatePeak(f[[4]], tssRegion=c(-1000, 1000), TxDb=txdb, addFlankGeneInfo=TRUE, flankDistance=5000)
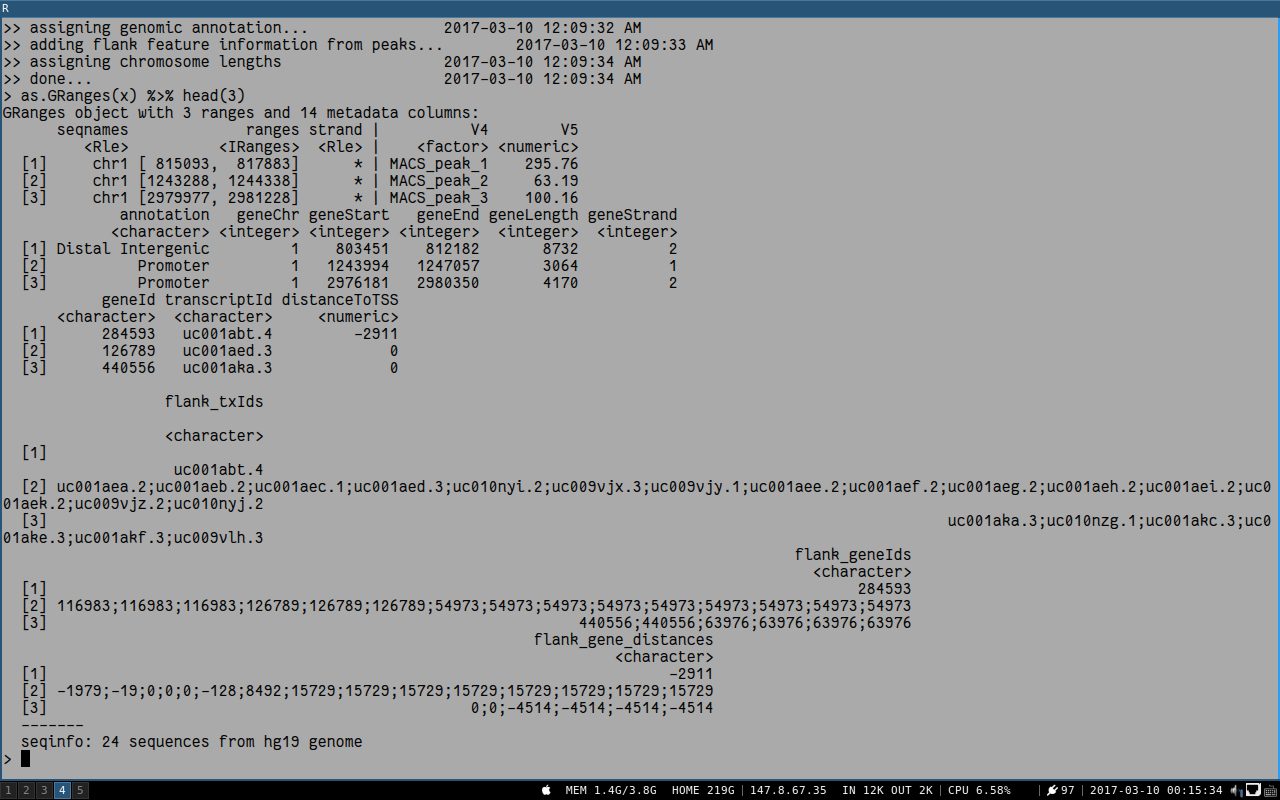
输出中多三列: flank_txIds, flank_geneIds和flank_gene_distances,在指定范围内所有的基因都被列出。
基因注释
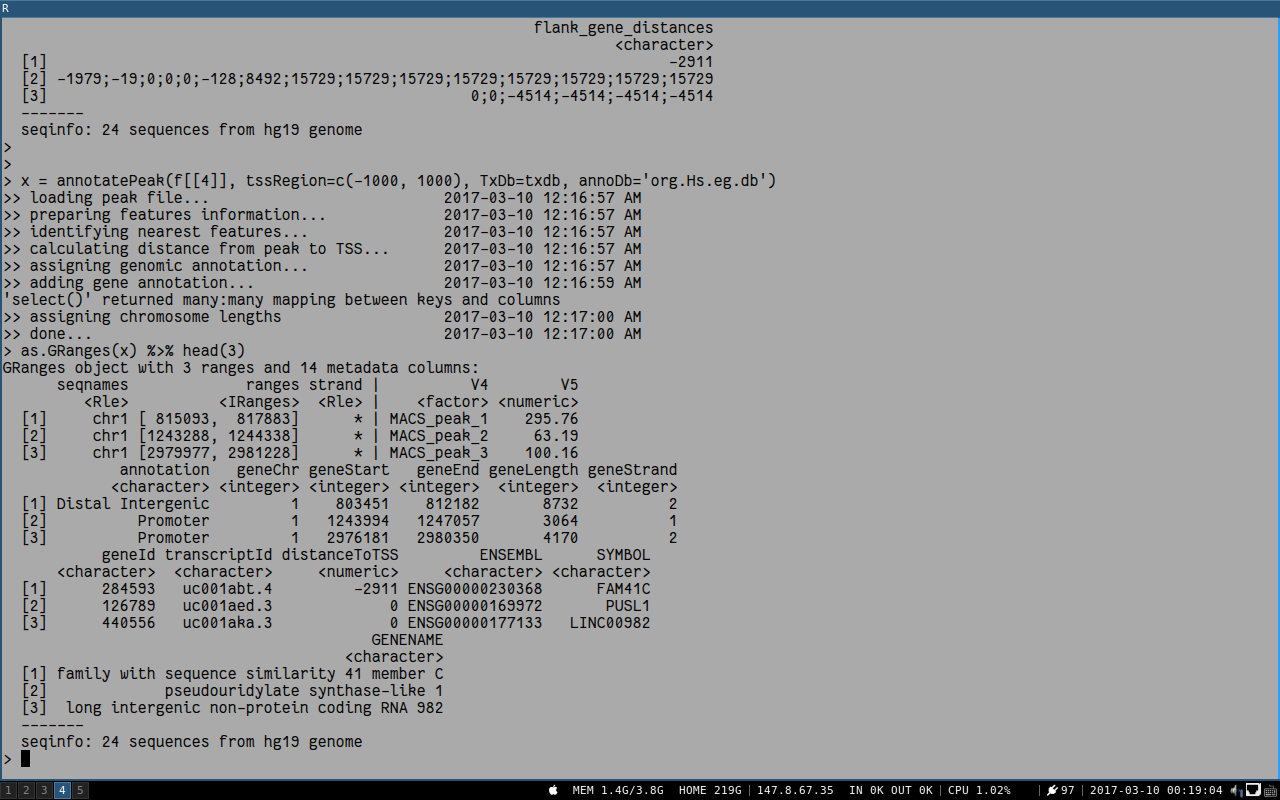
对于通常情况找最近基因的策略,最近基因给出来了,但都是各种人类不友好的ID,我们不能把一切都交给计算机,输出的结果我们还是要看一看的,能不能把基因的ID换成对人类友好的基因名,并给出描述性的全称,这个必然可以有。
只需要给annotatePeak传入annoDb参数就行了。
如果你的TxDb的基因ID是Entrez,它会转成ENSEMBL,反之亦然,当然不管是那一种,都会给出SYMBOL,还有描述性的GENENAME.
Citation
Yu G, Wang LG and He QY*. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31(14):2382-2383.


