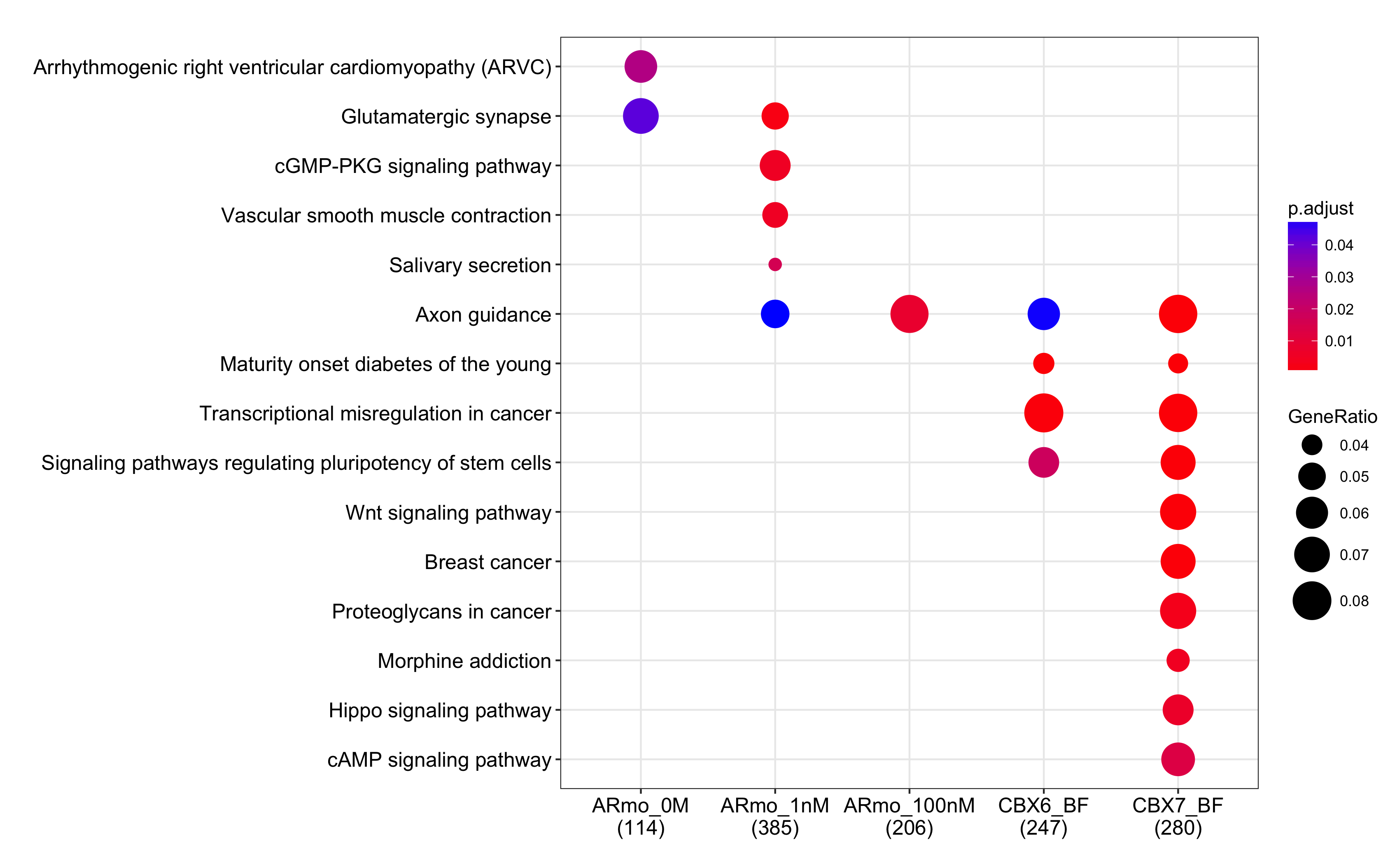
Genomic coordination的富集性分析
在《CS7:Genomic coordination的富集性分析(1)》说到了seq2pathway这个包,其实是两部曲,seq2gene->gene2pathway,无非是把测序片段用临近的基因注释,包括和TSS overlap的基因,宿主基因,上下游的基因等,然后拿这些基因跑ORA,做富集,仅此而已,这个包支持的物种极有限,《CS4:关于ChIPseq注释的几个问题》这一文中讲到ChIPseeker支持所有有基因组注释的物种,而《clusterProfiler for enrichment analysis》也支持所有物种(即使你自己跑的电子注释,也能支持),那么使用ChIPseeker来做基因注释,然后衔接clusterProfiler就可以支持所有物种的测序片段进行功能富集分析了。
《CS3: peak注释》本身就支持几种注释,另外我写了一个seq2gene的函数,套用seq2pathway的思路,把一个基因位置上所有关联的基因全部返回来,我们可以使用它去把基因位置信息转换成基因列表,然后用于富集分析,还是熟悉的味道,还是熟悉的配方🦄
library(ChIPseeker)
library(clusterProfiler)
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
bedfile=getSampleFiles()
seq=lapply(bedfile, readPeakFile)
genes=lapply(seq, function(i)
seq2gene(i, c(-1000, 3000), 3000, TxDb=txdb))
cc = compareCluster(geneClusters = genes,
fun="enrichKEGG", organism="hsa")
dotplot(cc, showCategory=10)