
眼看着2月14号就要到来了,又是一年一度的画红心装13的时候,这种事情已经被我BS了,请猛击:《画❤️装geek什么的,都弱爆了!😜》。红心已经没什么好画的了,特别是在我画了生日蛋糕,并用蛋糕画红心之后,《130岁了,祝你生快》,是驴子是马,都没法晒肌肉了。当然红心还是会有所谓的理工宅男晒,除此之外还会有各种花招,来给单身狗撒狗粮。想要加入撒狗粮大军的,快点看《如何告别单身》。
话说有一只蛙,它旅游去了广东,游戏结束。
这篇文章,只想跟大家讲一讲ggtree,你值得拥有的强大的进化树可视化软件,高颜值的图,就在于指尖几行代码中。
最近养蛙很流行,就以蛙为例,读树用read.tree,然后一个ggtree的指令,树就画出来了。而你见证神奇的时刻就在于你不断+geom_xxx加图层,这里我用了geom_tiplab来加taxa label,而这个可不仅仅是名字哦,用emoji也可以,这里我用的竟然是图片,你没看错,也可以。只要指定geom="image"就OK,那个本来要打的文本,就会被当成是文件名,然后读图片画图片,一切尽在我的ggimage包。
今天以ggplot2的一个坑来说一下,坑无处不在,防不胜防,你大可以试一下下面的代码:
> set.seed(123)
> require(ggplot2)
Loading required package: ggplot2
> rnorm(3)
[1] 0.8005543 1.1902066 -1.6895557
> set.seed(123)
> rnorm(3)
[1] -0.5604756 -0.2301775 1.5587083
在两次set.seed和rnorm之间,第一次因为加载了ggplot2,结果就不一样了!这必须是第二次是正确答案,也就是说加载ggplot2把你的seed给吃了!加载包会改变R环境?这绝对不是好主意,我们来试试加载别的包试试,比如我的clusterProfiler:
大家是否还记得我的《webinar录播 (2017-10-24):plotting tree + data》,这可以说是R画图最好的课程,PPT也分享给大家《ggtree直播PPT第一部分》和《ggtree直播PPT第二部分》。
我在PPT中,用了meme,发现没有R包可以做,于是我写了一个R包来做这个事情。
后来又写一个R包来实现字体的阴影效果:
有小伙伴说他要用gage这个包,因为可以选择sigmet这个index,然后得到的结果只有signaling and metabolic pathways,而不会有他不关心的disease pathways。然而也有各种不爽,他最喜欢的还是clusterProfiler,但没办法只做某些pathways。
我发现大家对clusterProfiler有各种误解,各种觉得没办法,我也很无语啊,明明我写了大量的文档,你们偏不看。clusterProfiler啥都可以做,你想做COG,domain这些没有内置支持的富集分析都可以的,因为clusterProfiler是通用的分析工具,啥都能做。
说到gage的pathway index,这其实是他们对pathway有个分类,这个数据就在https://pathview.uncc.edu/data/khier.tsv可以下载到,要支持他还不容易,但我不喜欢把别人的东西打包在自己的包里,所谓走别人的路,让别人无路可走,这可不是什么好主意。所以呢,我不会内置支持的,你们自己玩。
之前我发表读书笔记《主成分分析》
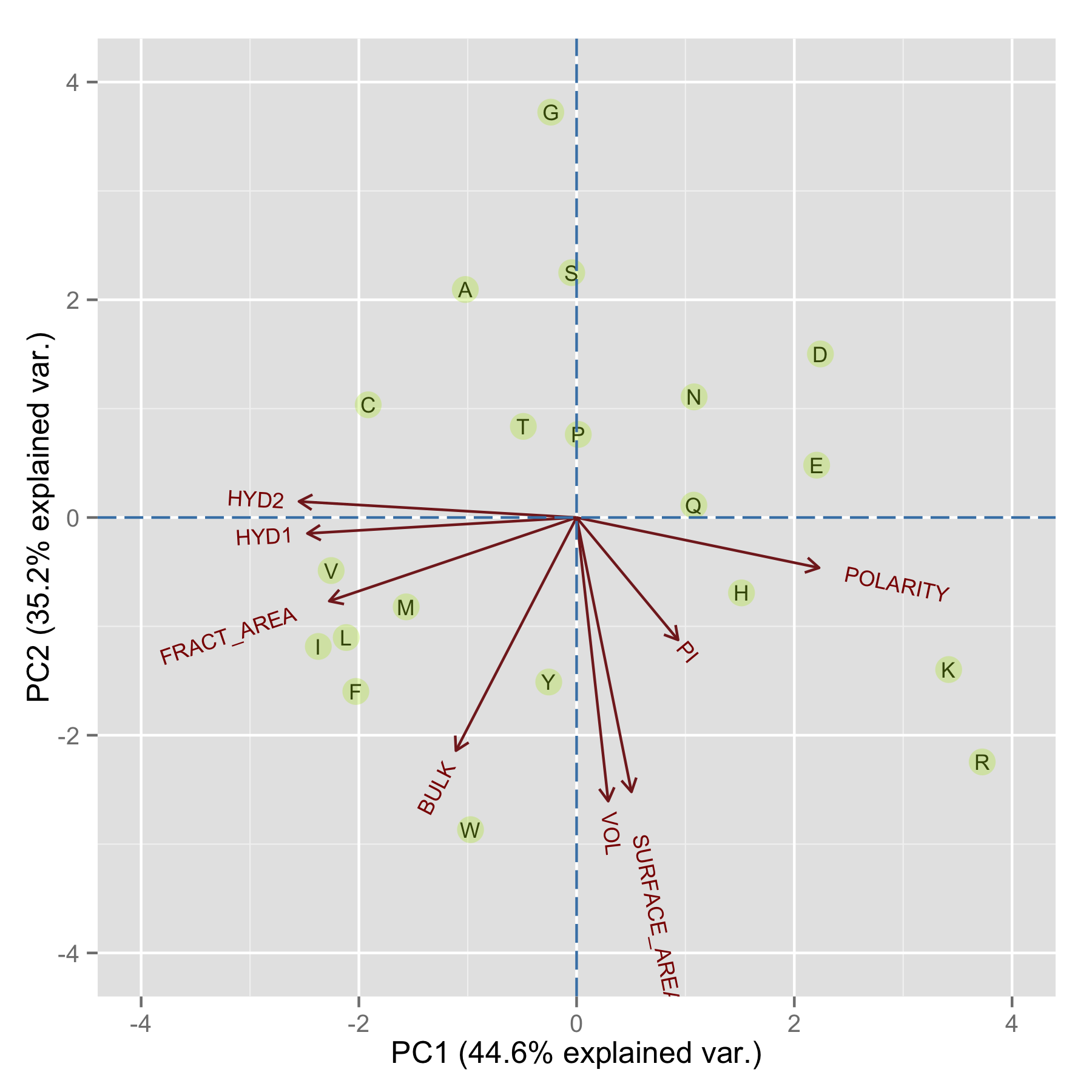
这可能是你见过最好看的PCA图了,有人在「宏基因组」群里问有没有什么包可以画?像这种提问,我以前是吐槽过的,请猛击《如何画类似MEME的注释序列》,当然说什么都没用,大家就是喜欢凡事问有包吗?因为包治百病嘛,不信你送个包给你女票试试!
jimmy回答说ggbiplot可以画差不多的,于是「宏基因组」公众号立刻就出来一篇《ggbiplot-最好看的PCA作图:样品PCA散点+分组椭圆+变量贡献与相关》,后面又有人提问,能不能加两个置信区间?@Chenhao童鞋就给出了解决方案,并且写了篇博客文介绍.
他的作图是基于ggord包,只能应用于LDA,于是「宏基因组」公众号又跟进发一篇《比PCA更好用的监督排序—LDA分析、作图及添加置信-ggord》,你会发现他们介绍同一类型的作图,一会这个包,一会那个包,一会某包有A功能,某包又有B功能,我就不说什么了。
很多人会有这样一个问题,差异基因一大堆,到底该选那个来做下游的实验验证?这个问题,说白了是个基因「重要性」的打分问题,你做差异基因分析的时候,就可以看做是个打分的过程,p值是你的统计量,p值越小,打分越高,然而所有的打分都是辅助帮我们进一步缩小范围而已,并不是打分越高就越「重要」,如果打分可以说明一切,那么我们就不需要实验验证了。所以像差异基因分析,我们一般是卡p < 0.01或p < 0.05来过滤,把p值小的基因留下来,但我们并不能说p值最小的基因就是最重要的。
回到开头的问题,我们要的是对打分(差异分析)结果再利用别的手段,再一次进行打分,进一步缩小范围。比如你可以通过构建相互作用网络,通过分析betweenness,找hub分子。当然打分不一定是要基于基因/蛋白水平,也可以是通路水平,比如你可以用clusterProfiler进行富集分析,然后把你的目标限定在某个/些通路上。反正就是各种手段一起上,直到你能够限定到少量几个基因上,对于做实验验证的人来说,再好不过。
上一周发表的《GOSemSim: GO语义相似性度量》,我记录了GOSemSim包被应用于各种各样的场景,它当然也可以拿来给基因/蛋白质打分。比如你用clusterProfiler分析后,就想验证某一通路,但不知道要选这个通路的哪个基因来做为切入点。
首先问一个问题,同一通路上的基因功能相似性高吗?大家可能会潜意识地认为应该比较高,这不一定的,基因/蛋白有直接或间接的相互作用,但这种相互作用可能只是「月上柳梢头,人约黄昏后」而已,可能偶尔才来一发,这种属于约会型。一个基因/蛋白通常会参与到多条不同的通路中,如果两个蛋白在不同的通路中经常一起出现,那么它们的功能相似性才会高,这种属于基友/闺蜜的死党型。今天就来讲一讲到底谁和谁在约会,谁和谁又是死党。
Y Han, G Yu, H Sarioglu, A Caballero-Martinez, F Schlott, M Ueffing, H Haase, C Peschel, AM Krackhardt. Proteomic investigation of the interactome of FMNL1 in hematopoietic cells unveils a role in calcium-dependent membrane plasticity. Journal of Proteomics. 2013, 78:72-82.
这篇文章是和慕尼黑工业大学(Technische Universität München)合作的一篇文章,使用了Co-IP去拉蛋白,再用LC-MS/MS进行鉴定,Co-IP是鉴定蛋白相互作用的常用手段,当然拉下来的蛋白不见得就是有真实的相互作用,它甚至于可能只是背景污染而已,所以我们需要对拉下来的蛋白进行打分,找出一些可能性比较高的候选蛋白进一步进行验证。


