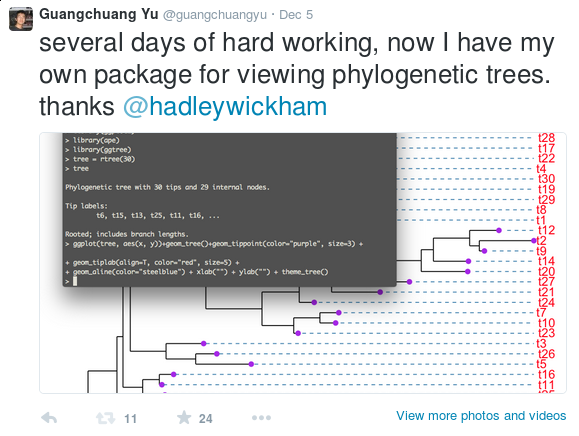
FigTree is designed for viewing
beast output as demonstrated by their example data:
clusterProfiler was used to visualize DAVID results in a paper published in BMC Genomics.

Some users told me that they may want to use DAVID at some circumstances. I think it maybe a good idea to make clusterProfiler supports DAVID, so that DAVID users can use visualization functions provided by clusterProfiler.
require(DOSE)
require(clusterProfiler)
data(geneList)
gene = names(geneList)[abs(geneList) > 2]
david = enrichDAVID(gene = gene, idType="ENTREZ_GENE_ID",
listType="Gene", annotation="KEGG_PATHWAY")
> summary(david)
ID Description GeneRatio BgRatio pvalue
hsa04110 hsa04110 Cell cycle 11/68 125/5085 4.254437e-06
hsa04114 hsa04114 Oocyte meiosis 10/68 110/5085 1.119764e-05
hsa03320 hsa03320 PPAR signaling pathway 7/68 69/5085 2.606715e-04
p.adjust qvalue geneID
hsa04110 0.0003998379 NA 9133/4174/890/991/1111/891/7272/8318/4085/983/9232
hsa04114 0.0005261534 NA 9133/5241/51806/3708/991/891/4085/983/9232/6790
hsa03320 0.0081354974 NA 4312/2167/5346/5105/3158/9370/9415
Count
hsa04110 11
hsa04114 10
hsa03320 7
There are only 5085 human genes annotated by KEGG, this is due to out-of-date DAVID data.
I am very exciting that I have received very positive feedback from Ahmed Moustafa and Simon Frost.
ggtree now has equipped with
a lot of new features. This time, I would like to introduce the replace
operator, %<%. Suppose we have build a tree view using ggtree with
multiple layers, we don’t need to run the code again to build a new tree
view with another tree. In
ggtree, we provides an operator, %<%, for updating tree view.
KEGG.db is not updated since 2012. The data is now pretty old, but many of the Bioconductor packages still using it for KEGG annotation and enrichment analysis. As pointed out in ‘Are there too many biological databases’, there is a problem that many out of date biological databases often don’t get offline. This issue also exists in web-server or software that using out-of-date data. For example, the WEGO web-server stopped updating GO annotation data since 2009, and WEGO still online with many people using it. The biological story may changed totally if using a recently updated data. Seriously, We should keep an eye on this issue.
Now enrichKEGG function is reloaded with a new parameter use_internal_data. This parameter is by default setting to FALSE, and enrichKEGG function will download the latest KEGG data for enrichment analysis. If the parameter use_internal_data is explicitly setting to TRUE, it will use the KEGG.db which is still supported but not recommended. With this new feature, supported species is unlimited if only there are KEGG annotations available in KEGG database. You can access the full list of species supported by KEGG via: http://www.genome.jp/kegg/catalog/org_list.html Now the organism parameter in enrichKEGG should be abbreviation of academic name, for example ‘hsa’ for human and ‘mmu’ for mouse. It accepts any species listed in http://www.genome.jp/kegg/catalog/org_list.html. In the current release version of clusterProfiler (in Bioconductor 3.0), enrichKEGG supports about 20 species, and the organism parameter accept common name of species, for instance “human” and “mouse”. For these previously supported species, common name is also supported. So that you script is still working with new version of clusterProfiler. For other species, common name is not supported, since I don’t want to maintain such a long mapping list with many species have no common name available and it may also introduce unexpected bugs.
I like running fortune every time when the terminal was started. A screenshot is shown below:
When I need to annotate nucleotide substitutions in the phylogenetic tree, I found that all the software are designed to display the tree but not annotating it. Some of them may support annotating the tree with specific data such as bootstrap values, but they are restricted to a few supported data types. It is hard/impossible to inject user specific data.