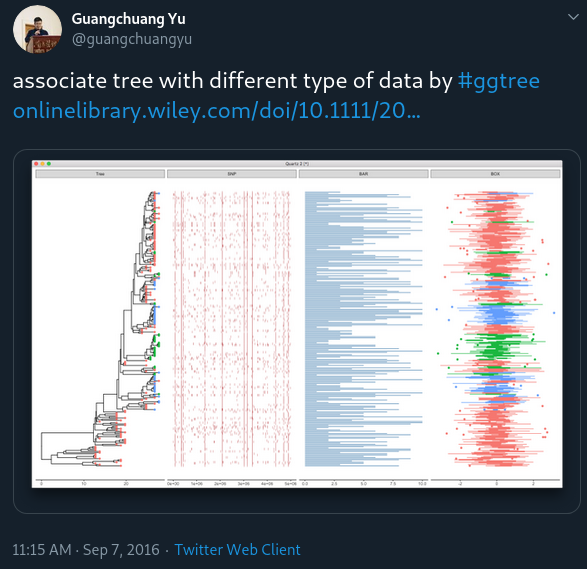
This is a question from ggtree google group:
Dear ggtree team,
how can I apply a geom_xxx to only one facet panel? For example if i want to get geom_hline(yintersect=1:30) or a geom_text() in the dot panel?
I cant see the facet_grid(. ~ var) function call, so I don’t know which subsetting to use. I have already read http://stackoverflow.com/questions/29873155/geom-text-and-facets-not-working
tr <- rtree(30)
d1 <- data.frame(id=tr$tip.label, val=rnorm(30, sd=3))
p <- ggtree(tr)
p2 <- facet_plot(p, panel="dot", data=d1, geom=geom_point, aes(x=val), color='firebrick')
d2 <- data.frame(id=tr$tip.label, value = abs(rnorm(30, mean=100, sd=50)))
p3 <- facet_plot(p2, panel='bar', data=d2, geom=geom_segment, aes(x=0, xend=value, y=y, yend=y), size=3, color='steelblue') + theme_tree2()
Thanks!
Andreas
If this can be done, we can create even more comprehensive tree plots.
Continue reading