I have been using clusterProfiler, which is a very useful package for gene set analysis and visualisation. I would like to use the ‘cnetplot’ function to plot a network of GO terms and the related genes. However for larger networks, the automatic display can be confusing and it would be helpful to be able to move nodes around. In the past I could do this with with cnetplot(fixed=FALSE) option, but after updating R and re-installing clusterProfiler, the output remains static.
I am using R 3.5.3 with clusterProfiler v3.10.1 which I installed using Bioconductor 3.8. I have installed and loaded the ‘igraph’ package, and the following test code produces output in an interactive window, as desired:
library(igraph) g <- make_ring(10) tkplot(g)
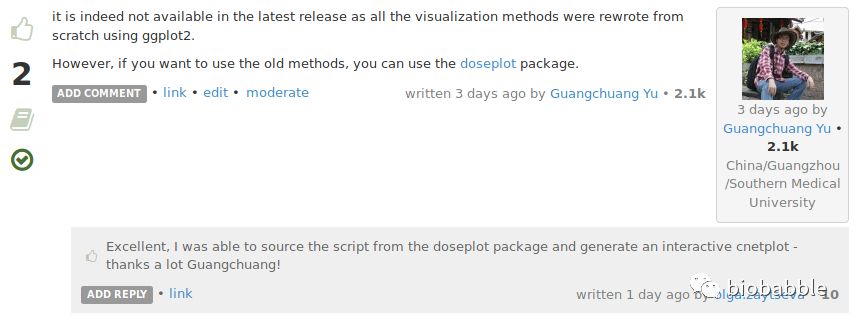
Is there any way to make cnetplot output interactive, or is that functionality simply not available in the latest release?
Any help would be greatly appreciated!