
I am very glad that ggtree is now available via Bioconductor. This is my 6th Bioconductor package. ggtree now supports parsing output files from BEAST, PAML, HYPHY, EPA and PPLACER and can annotate phylogenetic tree directly using plot methods.
I like running fortune every time when the terminal was started. A screenshot is shown below:
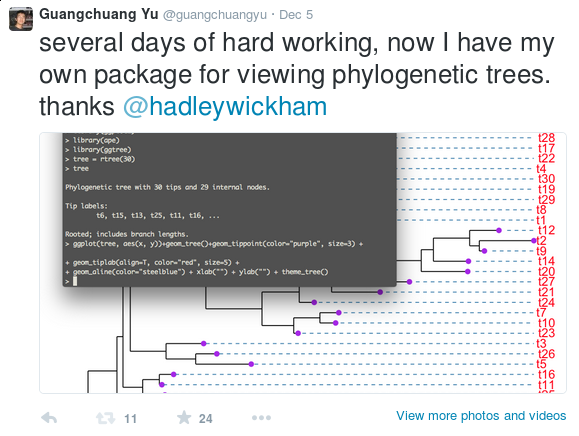
When I need to annotate nucleotide substitutions in the phylogenetic tree, I found that all the software are designed to display the tree but not annotating it. Some of them may support annotating the tree with specific data such as bootstrap values, but they are restricted to a few supported data types. It is hard/impossible to inject user specific data.
One day, I am looking for R packages that can analyze PPI and after searching, I found the ppiPre package in CRAN.

The SIR model divides the population to three compartments: Susceptible, Infected and Recovered. If the disease dynamic fits the SIR model, then the flow of individuals is one direction from the susceptible group to infected group and then to the recovered group. All individuals are assumed to be identical in terms of their susceptibility to infection, infectiousness if infected and mixing behaviour associated with disease transmission.
We defined: $S_t$ = the number of susceptible individuals at time t
$ I_t $ = the number of infected individuals at time t
$R_t$ = the number of recovered individuals at time t
Suppose on average every infected individual will contact $\gamma$ person, and $\kappa$ percent of these $\gamma$ person will be infected. Then on average there are $\beta = \gamma \times \kappa$ person will be infected an infected individual.
Nearest gene annotation
Almost all annotation software calculate the distance of a peak to the nearest TSS and assign the peak to that gene. This can be misleading, as binding sites might be located between two start sites of different genes or hit different genes which have the same TSS location in the genome.
The function annotatePeak provides option to assign genes with a max distance cutoff and all genes within this distance were reported for each peak.
Genetic drift is the term used in population genetics to refer to the statistical drift over time of gene frequencies in a population due to random sampling effects in the formation of successive generations. In a narrower sense, genetic drift refers to the expected population dynamics of neutral alleles (those defined as having no positive or negative impact on reproductive fitness), which are predicted to eventually become fixed at zero or 100% frequency in the absence of other mechanisms affecting allele distributions.
The most important keyword in the definition of genetic drift is random sampling effects. The figure belowed illustrates this idea. The surviving individuals do not necessarily have selection advantage. They are randomly selected.
