
BED文件
BED的全称是Browser Extensible Data,顾名思义是为genome browser设计的,大名鼎鼎的bedtools可不是什么「床上用品」。
BED包含有3个必须的字段和9个可选字段。
三个字段包括:
- 1 chrom - 染色体名字
- 2 chromStart - 染色体起始位点
- 3 chromEnd - 染色体终止位点
这里必须指出的是chromStart是起始于0,而不是1。很多分析软件都忽略 了这一点,会有一个碱基的位移,据我所知Homer和ChIPseeker没有这个问题,而像peakAnalyzer, ChIPpeakAnno等都有位移的问题。
可选的9个字段包括:
- 4 name - 名字
- 5 score - 分值(0-1000), 用于genome browser展示时上色。
- 6 strand - 正负链,对于ChIPseq数据来说,一般没有正负链信息。
- 7 thickStart - 画矩形的起点
- 8 thickEnd - 画矩形的终点
- 9 itemRgb - RGB值
- 10 blockCount - 子元件(比如外显子)的数目
- 11 blockSizes - 子元件的大小
- 12 blockStarts - 子元件的起始位点
一般情况下,我们只用到前面5个字段,这也是做peak calling的MACS输出的字段。
其中第5个字段,MACS的解释是这样子的:
The 5th column in this file is the summit height of fragment pileup.
是片段堆积的峰高,这也不难理解,为什么我在ChIPseeker是画peak coverage的函数covplot要有个*weightCol*的参数了。
数据可视化
从名字上看,它是为genome browser而生,相应的,ChIPseeker实现了*covplot*来可视化BED数据。
covplot支持直接读文件出图:
library(ChIPseeker)
library(ggplot2)
files <- getSampleFiles()
covplot(files[[4]])
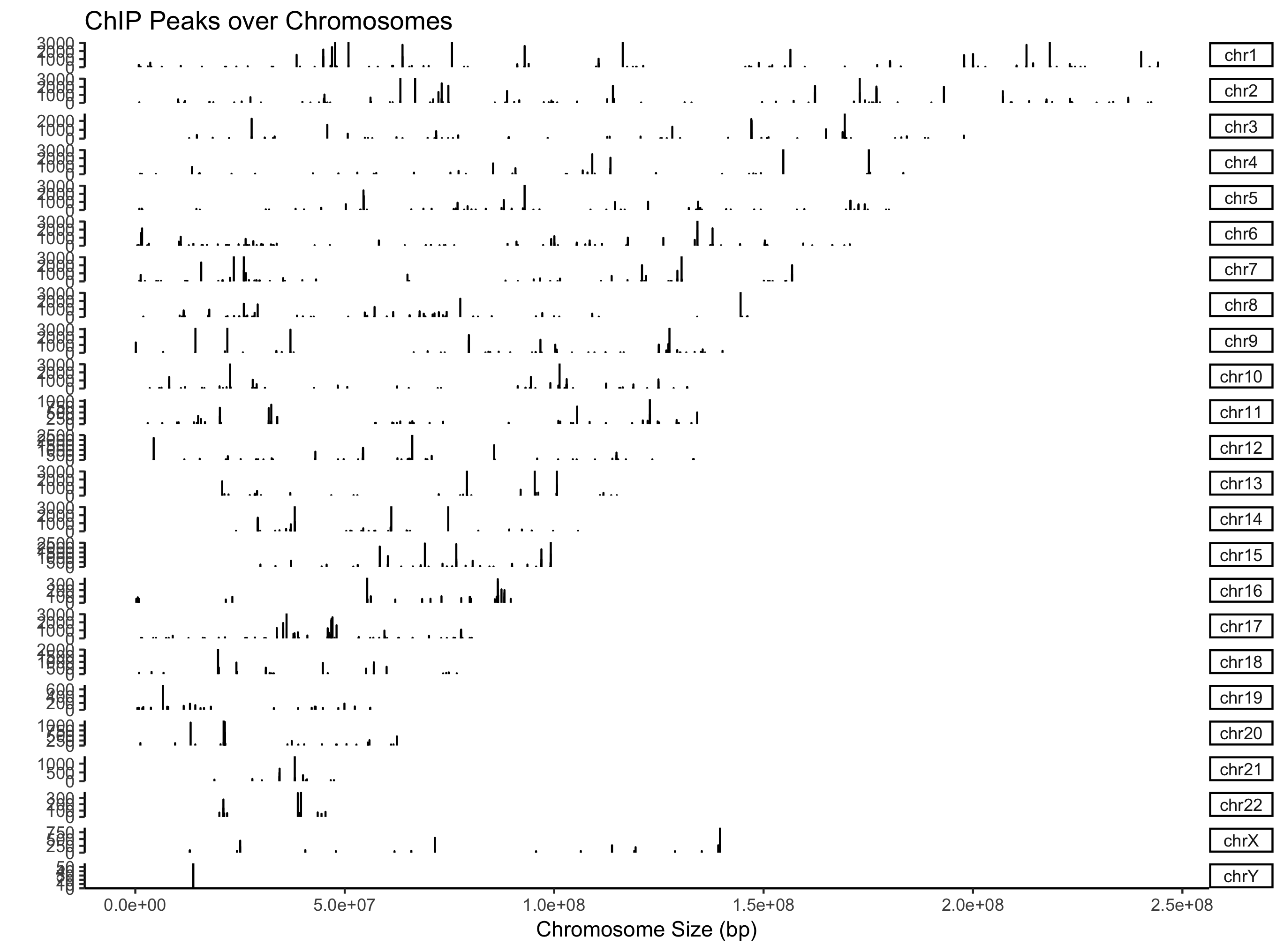
支持GRanges对象,同时可以多个文件或者GRangesList
peak=GenomicRanges::GRangesList(CBX6=readPeakFile(files[[4]]),
CBX7=readPeakFile(files[[5]]))
covplot(peak, weightCol="V5") + facet_grid(chr ~ .id)
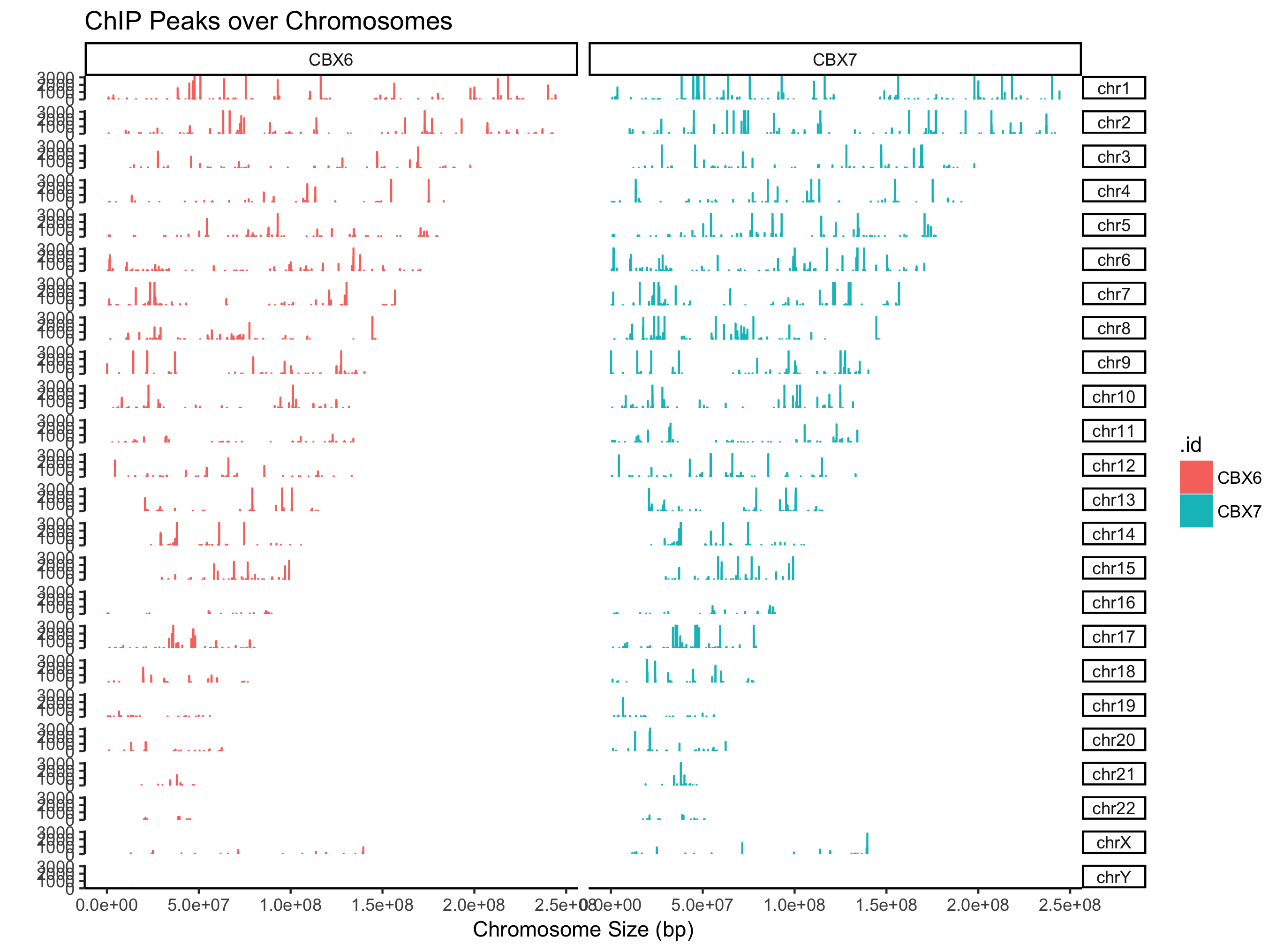
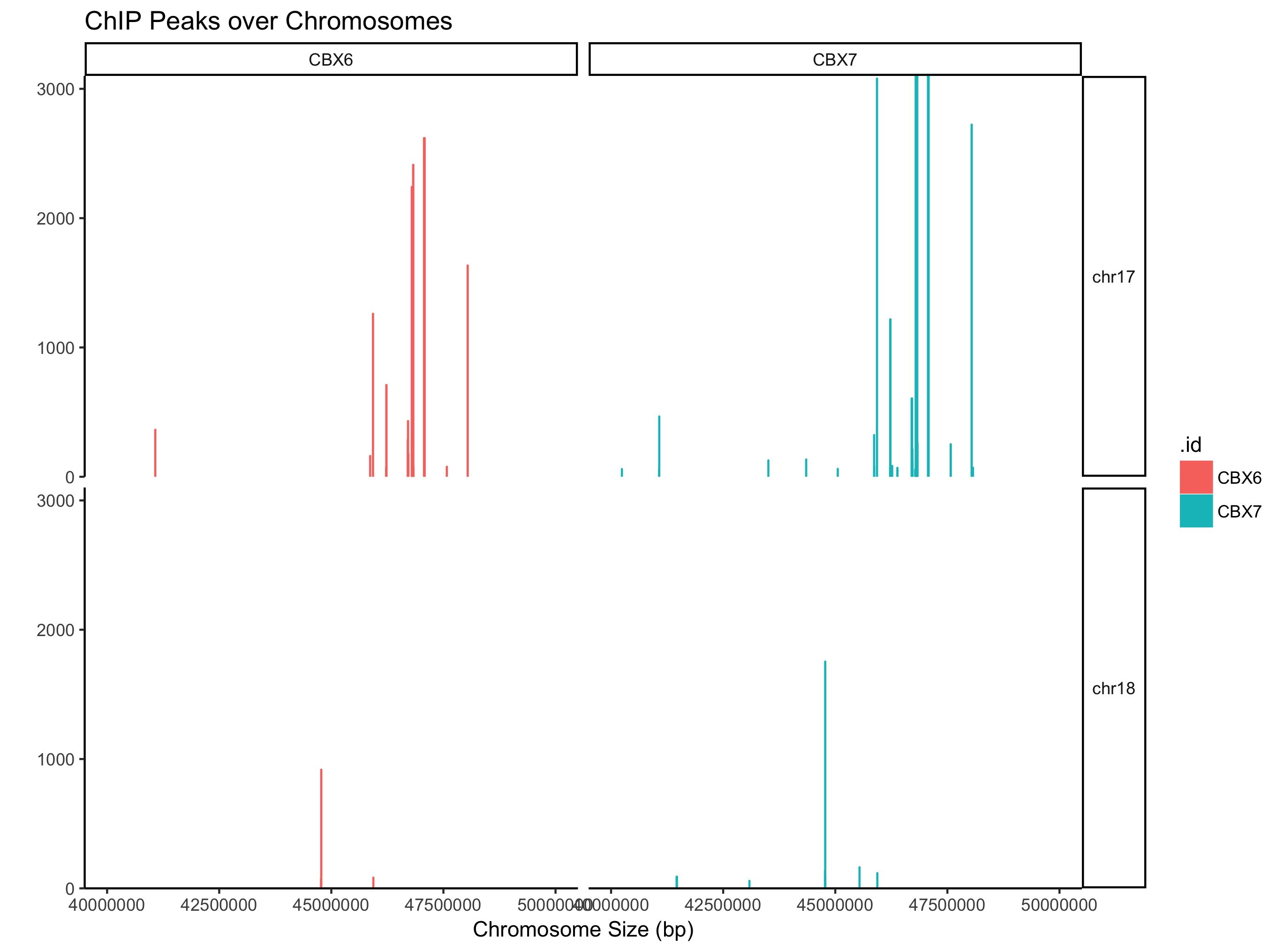
支持可视化某个窗口
covplot(peak, weightCol="V5", chrs=c("chr17", "chr18"), xlim=c(4e7, 5e7)) + facet_grid(chr ~ .id)
拿到数据后,我们首先会可视化看一下数据,接下来就会想知道这些peak都和什么样的基因有关,这将在下次讲解,如何做注释。
Citation
Yu G, Wang LG and He QY*. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31(14):2382-2383.


