
CS6: ChIP数据可视化
2017眼看要结束,立下写《CS0: ChIPseq从入门到放弃》的flag还没完成,当时ChIPseeker是33个引用,现在已经80了,时间过得好快。
最近放羊的Jimmy给我发来了一个截屏:
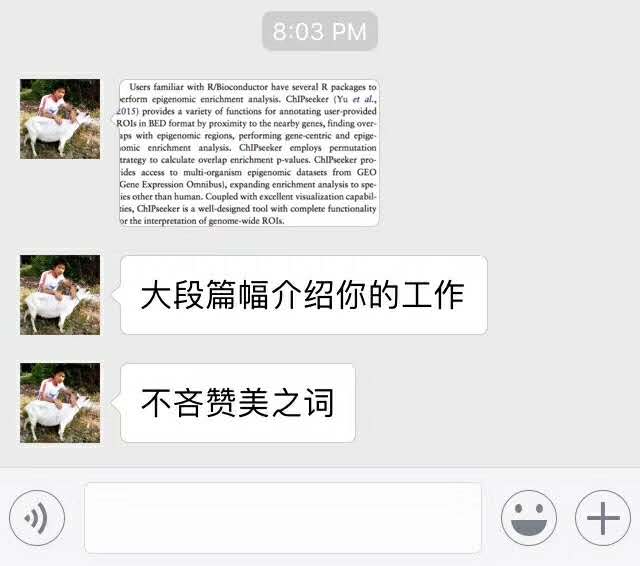
说了一篇新文章,大段在称赞ChIPseeker:

这篇文章的作者以前就跟我要ChIPseeker的文章,说是上课要给学生读,所以我要感谢他,上课和文章都给我打了广告!
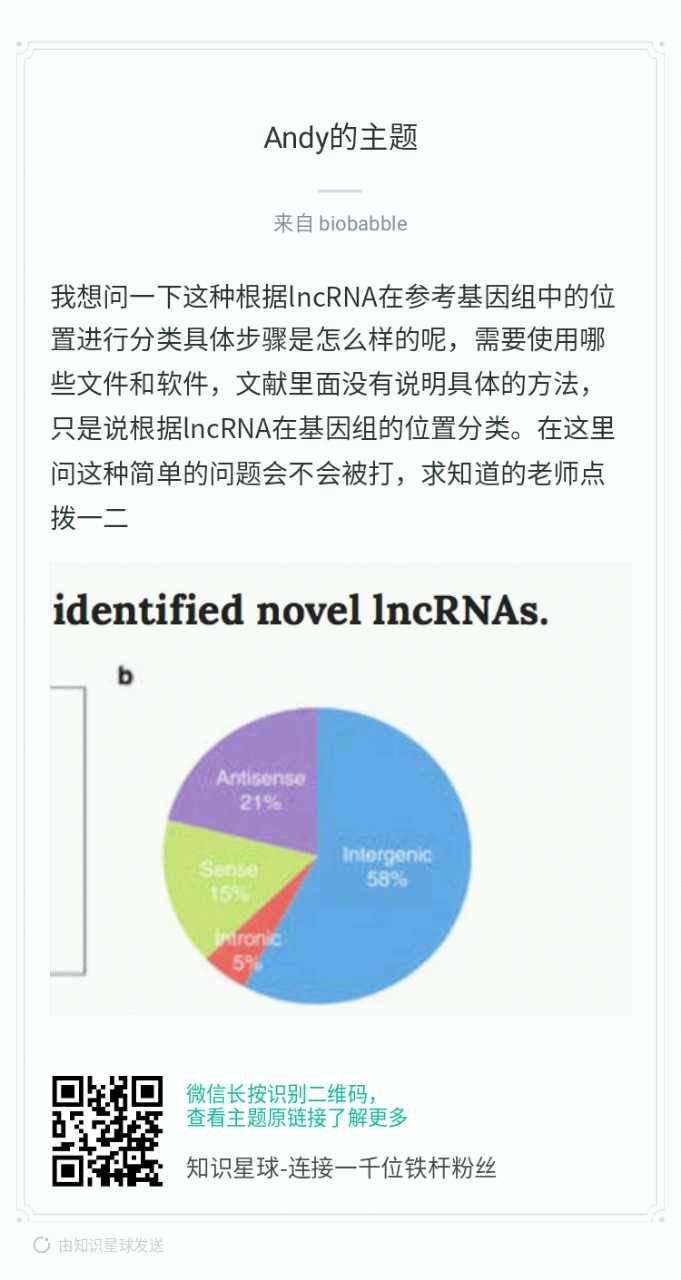
最近小密圈有小伙伴问怎么对lncRNAs按基因位置进行分类,我让他去看这个ChIPseeker系列文章,不要被ChIPseeker这个名字给蒙蔽了,我当年写这个包的时候,我只是想做ChIPseq,我也没想到自己写的是个通用的包,其实应该叫genomic coordinates annotation, visualization and comparison。ChIPseeker不单用于ChIPseq,所有genomic coordination通杀。在《CS4:关于ChIPseq注释的几个问题》里我就谈到了有人做DNA breakpoints的断点位置注释。在biostars上我也展示过注释lincRNA (https://www.biostars.org/p/215069/)。另外CS4里也提到了背景注释可以用GRanges对象,这保证了各种需求都可以满足。
如果你用ChIPseeker做注释,那么出上面的饼图,太容易,ChIPseeker只会做得更好。
Peak可视化
首先在分析之前,我们拿到BED文件,就可以先看看peak在整个染色体上的分布,这个在《CS2: BED文件 》一文里有展示,这里不在重复。
另外我们可以限定在一个固定的窗口里,然后把这些peak全部align起来,看看在某个窗口上的结合谱图。比如说启动子区域,使用转录起始位点,然后指定上下游,使用getPromoters函数,就可以为我们准备好窗口。再使用getTagMatrix函数,把peak比对到这个窗口,并生成矩阵,供我们分析、可视化。
library(ChIPseeker)
files <- getSampleFiles()
peak <- readPeakFile(files[[4]])
promoter <- getPromoters(TxDb=txdb,
upstream=3000, downstream=3000)
tagMatrix <- getTagMatrix(peak, windows=promoter)
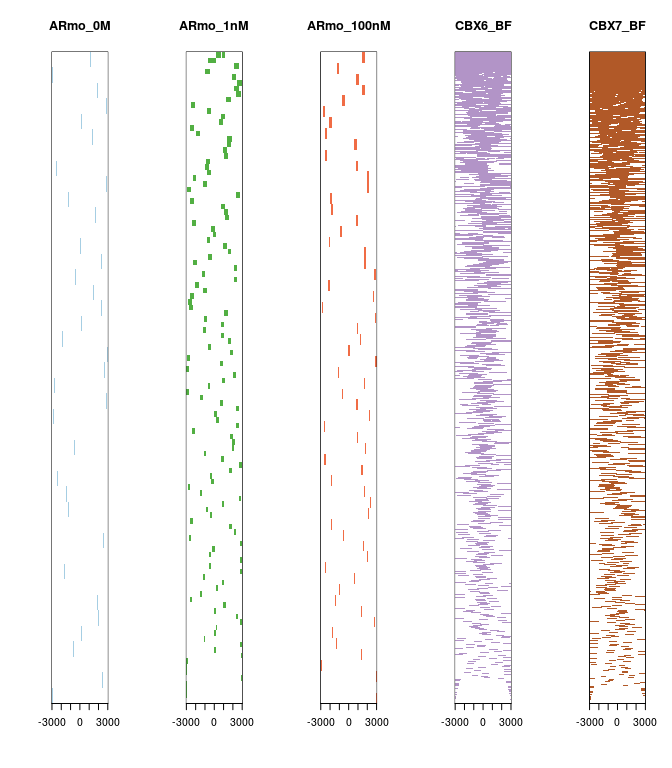
这时候,我们可以用tagHeatmap来画热图,颜色随意指定:
tagHeatmap(tagMatrix, xlim=c(-3000, 3000), color="red")

另外你可以直接从文件出发,ChIPseeker提供了peakHeatmap函数,你给文件名,就可以直接出图,两个函数都支持多种数据同时展示,方便比较,这里可以看出前三个样品结合位点不在转录起始区域,而后两个则是。
require(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
peakHeatmap(files, weightCol="V5", TxDb=txdb,
upstream=3000, downstream=3000,
color=rainbow(length(files)))
另外我们还可以以谱图的形式来展示结合的强度:
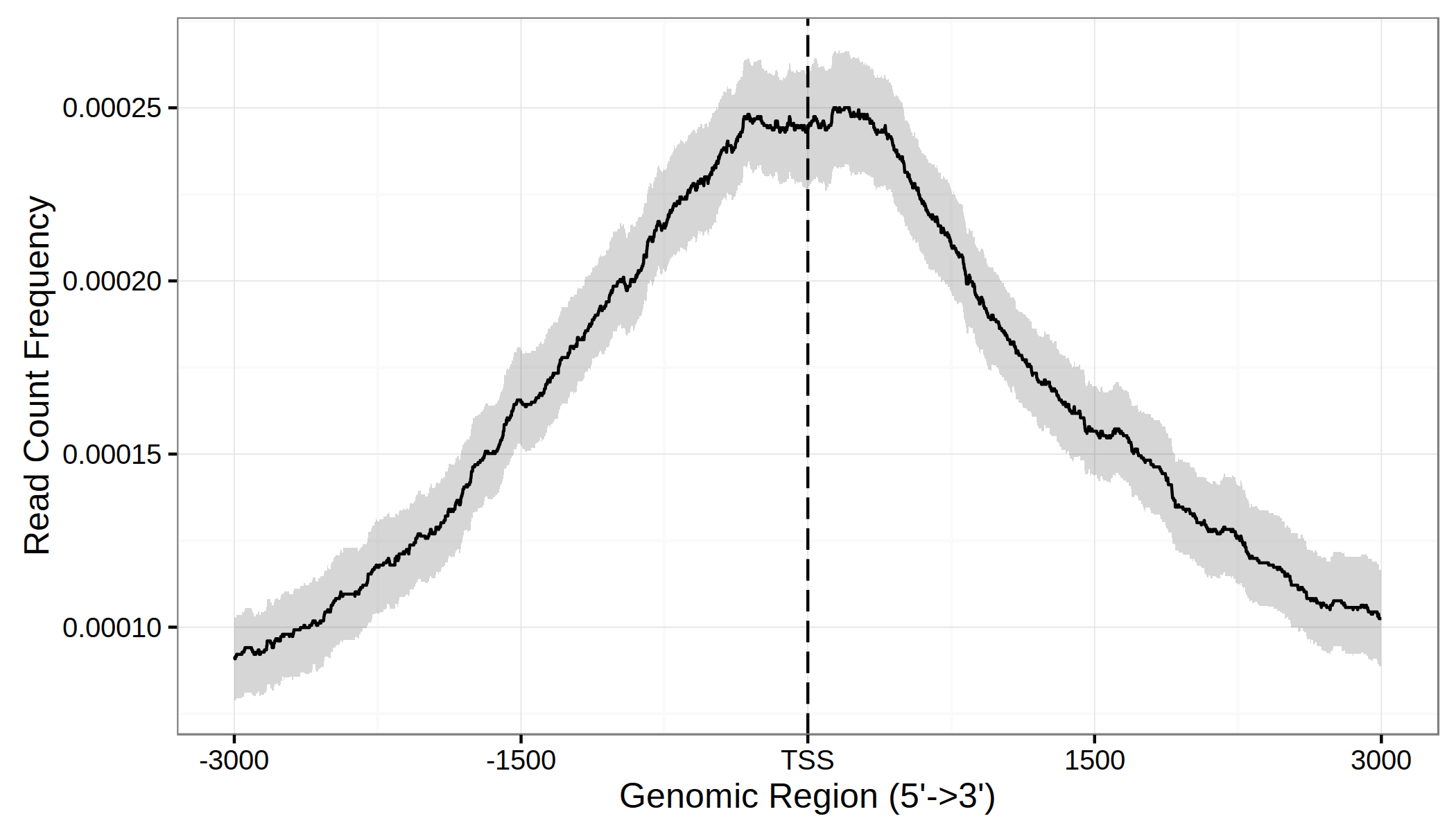
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency")
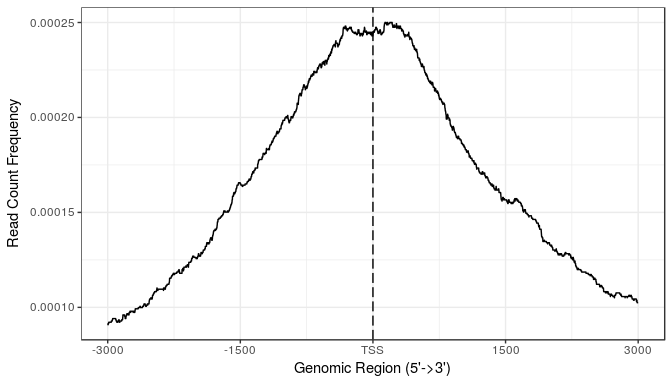
上面这个图可以非常清晰地看到,这个结合位点是集中在转录起始位点的。另外ChIPseeker也提供了plotAvgProf2函数,支持以文件为输入一步做图。下面这个代码,会产生上面一样的图。
plotAvgProf2(files[[4]], TxDb=txdb,
upstream=3000, downstream=3000,
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency")
另外这个函数还支持使用bootstrap估计置信区间:
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
conf = 0.95, resample = 1000)
这个功能由微博@Puriney贡献,你们可以去微博勾搭调戏,是个颜值很高的鲜肉。
同样这个函数也支持多个数据的比较:
tagMatrixList <- lapply(files, getTagMatrix,
windows=promoter)
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000))
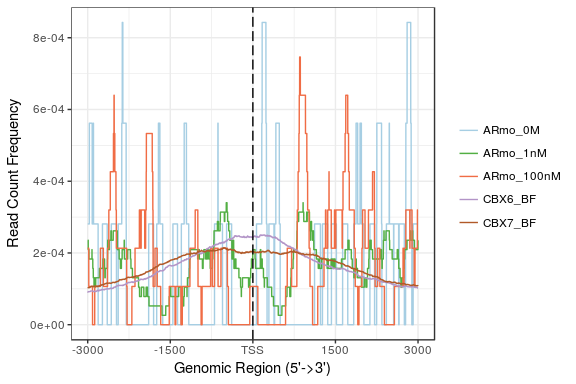
我们可以对其进行分面展示,当然也支持估计置信区间:
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000),
conf=0.95,resample=500, facet="row")
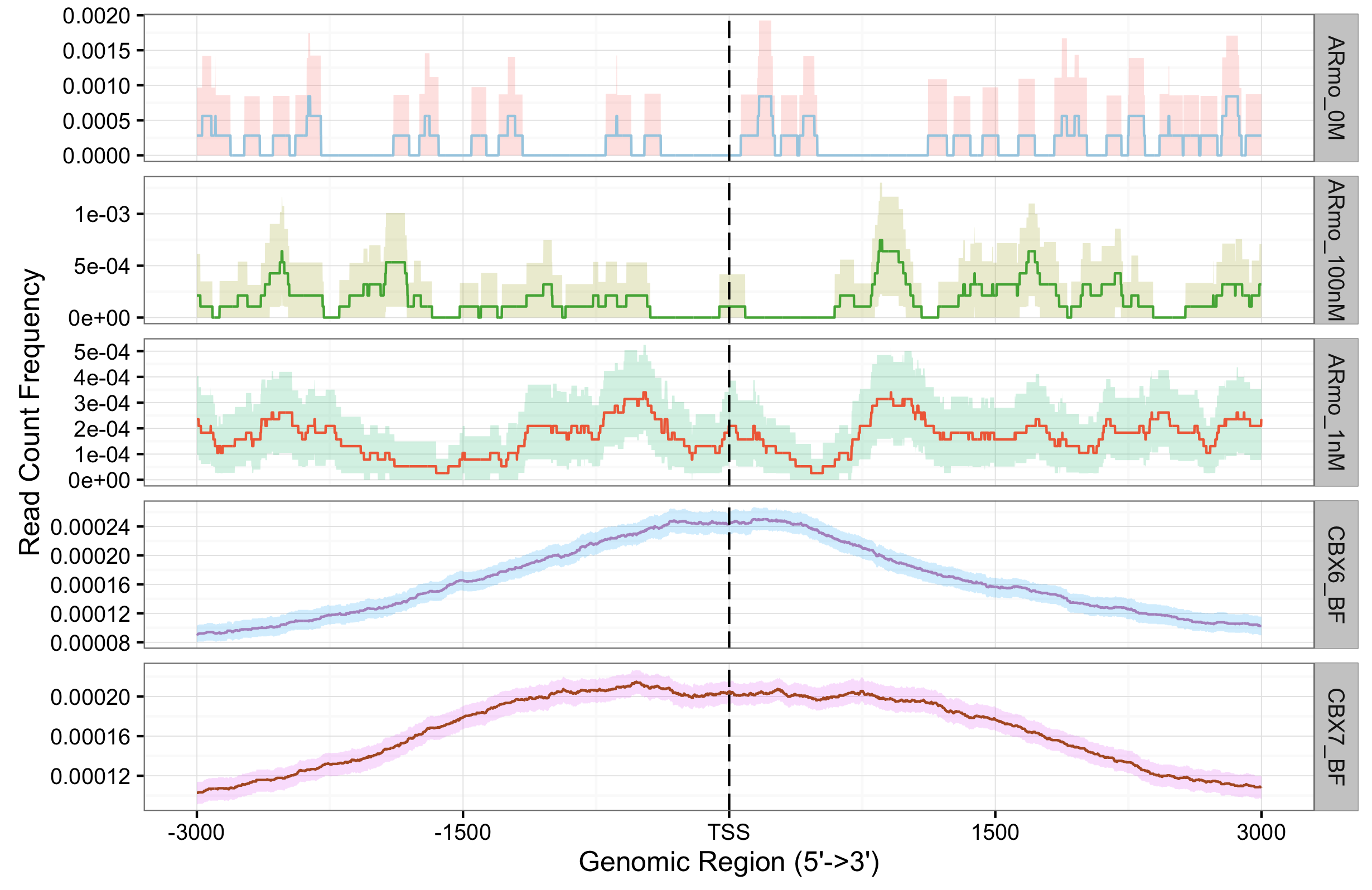
这和前面的热图是一致的,前三个样品显然不是调控转录的。
这里我展示的都是启动子窗口,然而ChIPseeker支持的并不止是启动子,不单单是看转录调控,比如说你是研究可变剪切的,那么蛋白结合到外显子/内含子的起始位置,显然就更感兴趣了。所以除了getPromoters函数之外,ChIPseeker还提供了getBioRegion函数,你可以指定’exon’或’intron’,它会像getPromoters一样为你准备好数据,即使这不是你感兴趣的,你也依然可以用它来准备窗口,然后可视化看一下,或许你能意外地发现你研究的蛋白竟然在外显子/内含子起始位置上有很强的结合谱,做为前期data exploration,「妹妹你大胆地往前走啊」,尽管啥都试试!
注释信息可视化
我在《CS3: peak注释》和《CS4:关于ChIPseq注释的几个问题》两篇文章里,重点讲了注释,注释后的信息当然也需要我们可视化展示,方便解析结果。
peakAnno <- annotatePeak(files[[4]], tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Hs.eg.db")
plotAnnoPie(peakAnno)
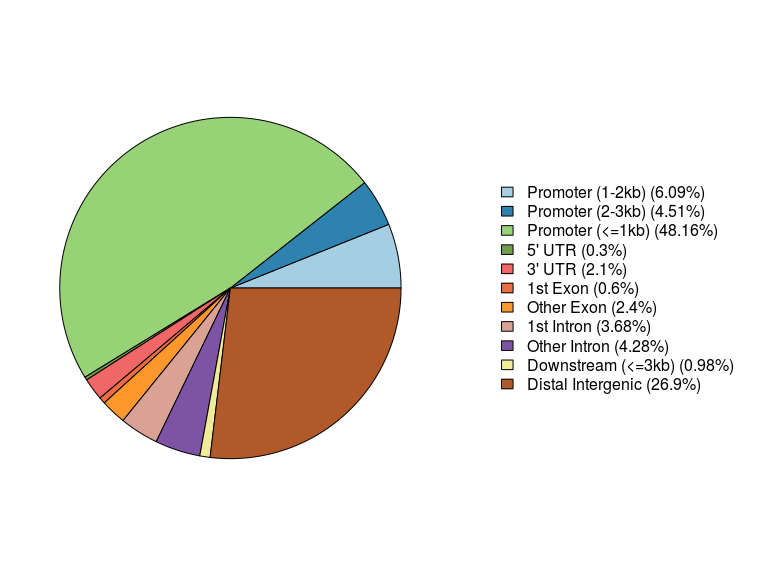
首先是饼图,这也是小密圈的小伙伴问的,peak落在了什么地方,启动子?外显子?内含子?还是基因间区?饼图给出了比例,当然一个peak所在的位置可能是一个基因的外显子而同时又是另一个基因的内含子,所以annotatePeak有个参数genomicAnnotationPriority让你指定优先顺序,默认顺序如下:
- Promoter
- 5’ UTR
- 3’ UTR
- Exon
- Intron
- Downstream
- Intergenic
这里有下游但没有上游,因为Promter我们定义为转录起始位点的上下游区域,所以它包括了上游,而下游就是在基因间区里,而我又把它单独拿出来,因为这是紧接着基因的下游,和远程的间区分开,通常默认参数就可以。
有饼图,当然有柱状图,这是躺平任调戏的柱子:
plotAnnoBar(peakAnno)
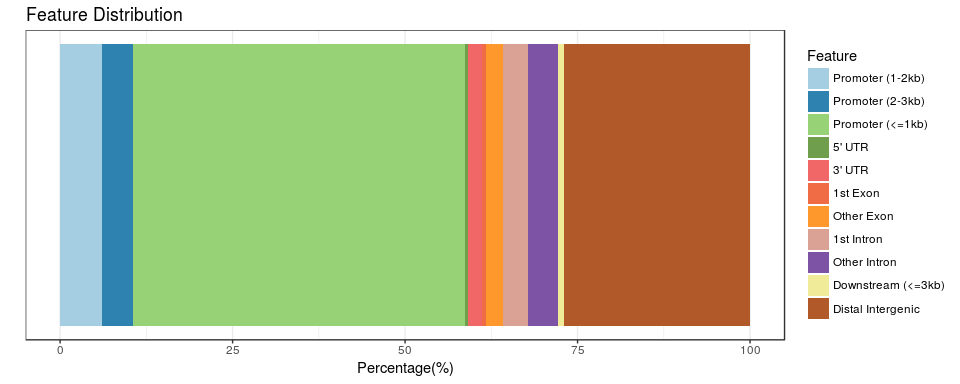
正如上面说的,一个结合位点可以是一个基因的Exon和另一个基因的Intron,除了按照优先级别给出唯一的注释之外,ChIPseeker其实对所有的注释都有记录,我当时就想怎么样把这些信息给展示出来,当时在笔记本上涂鸦了这样的图:
这个图我称之为vennpie取维恩图venn plot画交集和饼图pie plot画分布的结合。
vennpie(peakAnno)
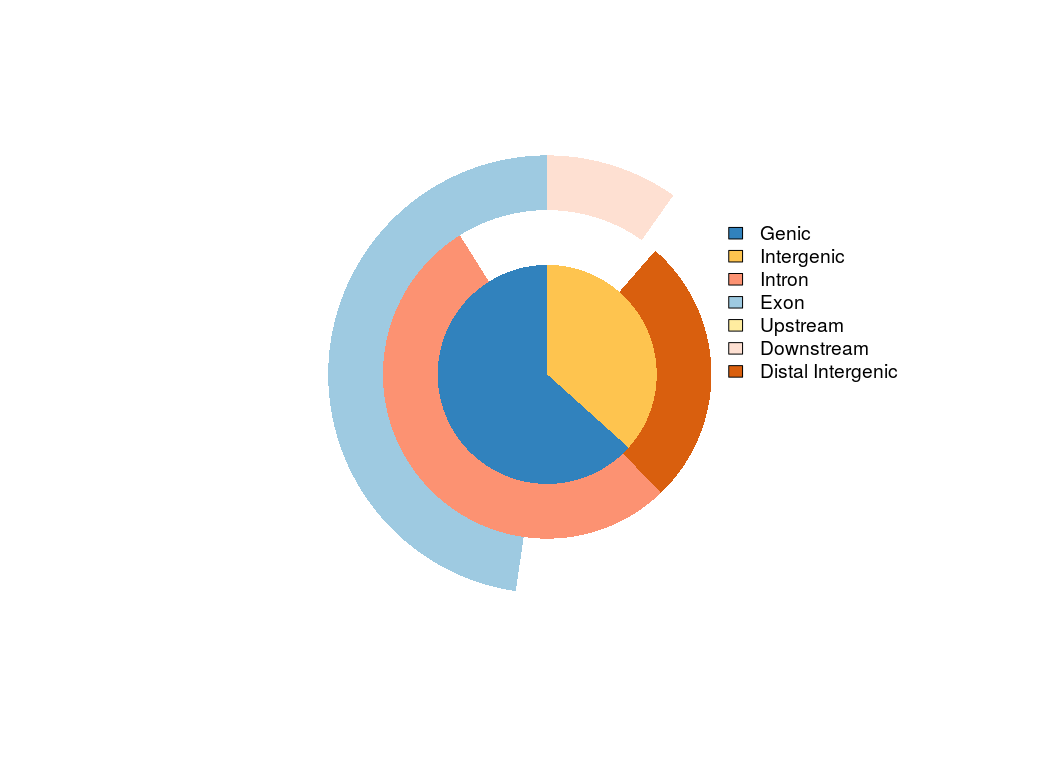
这个图讲真我很喜欢,但不满意,因为无法展示全部的信息。所以当我看到有UpSetR这个包的时候,我立刻就把它应用于展示注释信息的交集:
upsetplot(peakAnno)
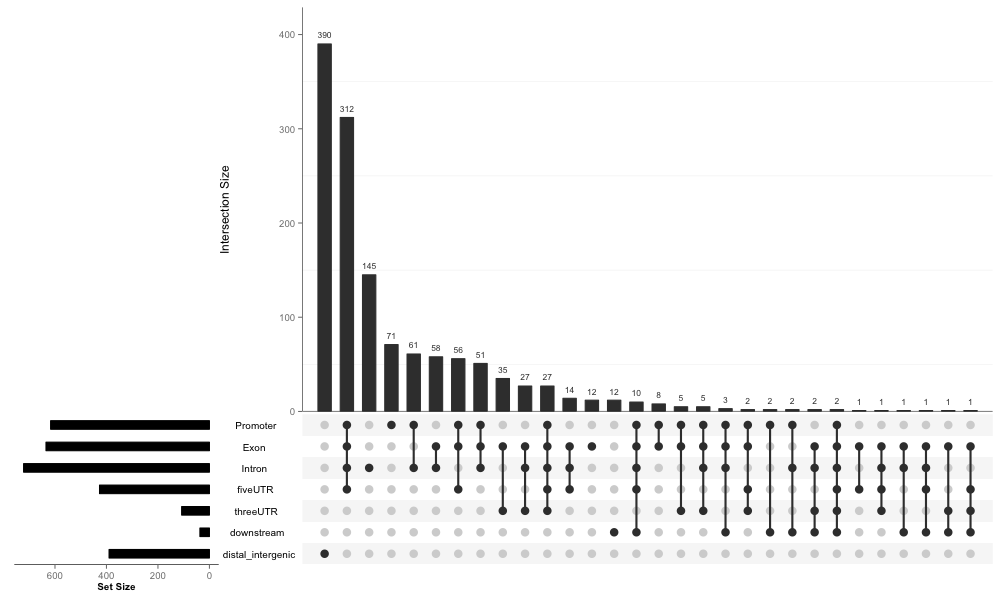
这个可以很好的把信息全部展示出来,然而第一不够vennpie直观,第二右上角空白太多,所以我就想把vennpie强插上去,最终通过参数实现这个效果:
upsetplot(peakAnno, vennpie=TRUE)
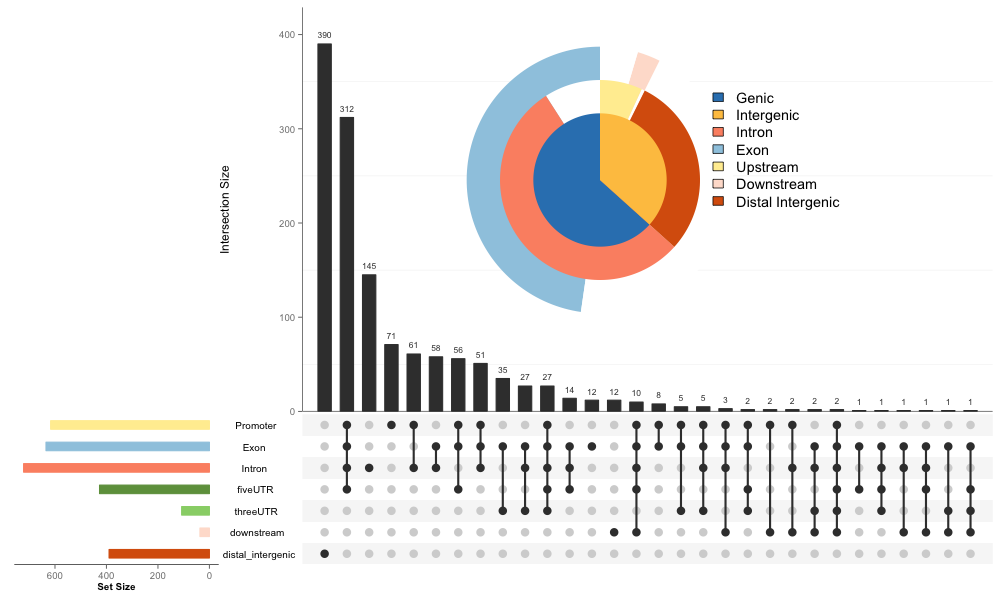
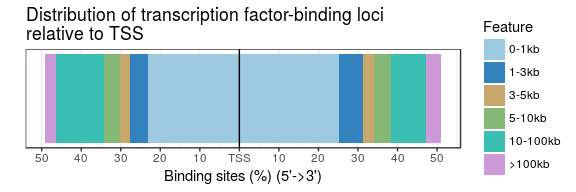
ChIPseeker还注释了最近的基因,peak离最近基因的距离分布是什么样子的?ChIPseeker提供了plotDistToTSS函数来画这个分布:
plotDistToTSS(peakAnno,
title="Distribution of transcription factor-binding loci\nrelative to TSS")
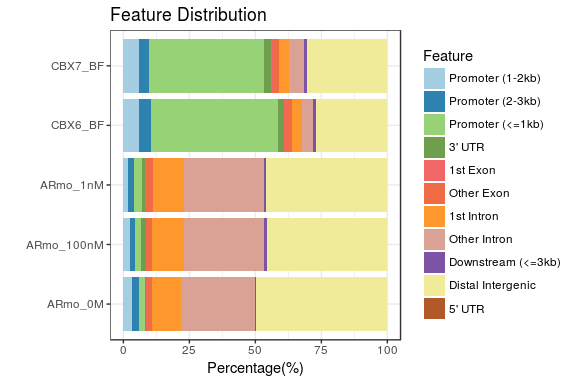
plotAnnoBar和plotDistToTSS这两个柱状图都支持多个数据同时展示,方便比较,比如:
peakAnnoList <- lapply(files, annotatePeak, TxDb=txdb,
tssRegion=c(-3000, 3000), verbose=FALSE)
plotAnnoBar(peakAnnoList)
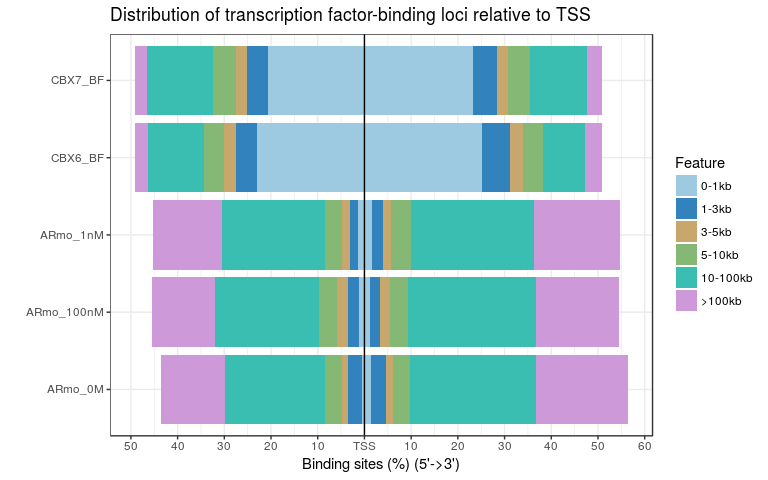
plotDistToTSS(peakAnnoList)
ChIPseeker还提供了一个vennplot函数,比如我想看注释的最近基因在不同样本中的overlap:
genes <- lapply(peakAnnoList, function(i)
as.data.frame(i)$geneId)
vennplot(genes[2:4], by='Vennerable')
如果你们有画图的需求,尽管来找我合作,有偿做图也可以,请参考「生信技能树」公众号的有偿专区。


