我这个公众号不像大多数所谓的生信从入门到精通的各种其实只是搬运点入门教程的群众喜闻乐见的公众号。正如我在《为什么要开这个公众号》里说的,这是小众的,有个人色彩的各种原创文。我不可能像其它公众号一样招两小弟当客服,很多人在公众号后台向我扔了许多问题,由于个人精力有限,只能优先解答「知识星球」的问题,上次写的《同一数据多变量分组的boxplot?》,图虽然简单,却穿着好多件马甲,而我把它扒光了给你看🙈
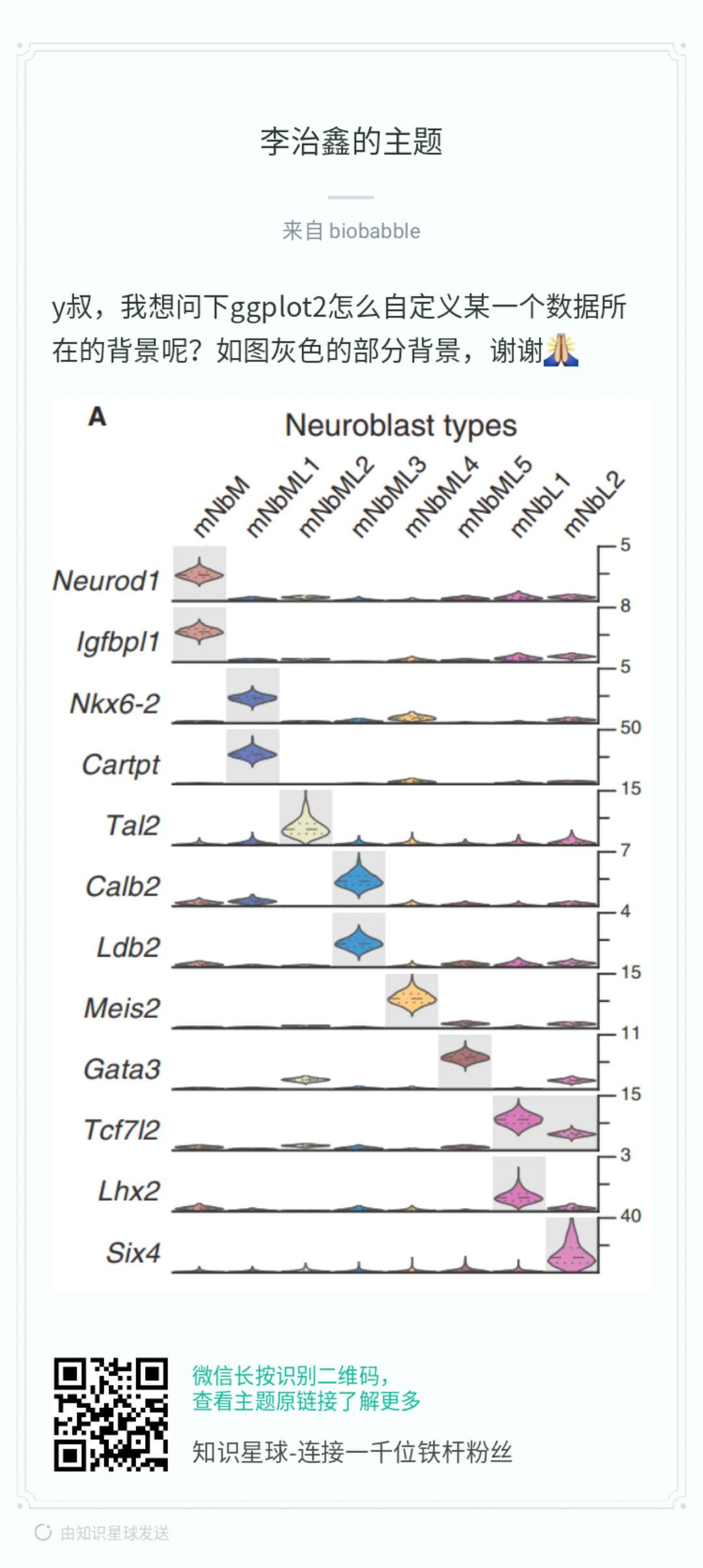
上面这个图,你看着高大上吧,我都可以吐它一脸口水。每一个有灰色背景的图,在x轴上violin都够到边界了,其实所有的violin都够到了,这证明什么?每个violin之间其实不可比较!你能想像几个独立的数据,在统一的bin width情况下,画density curve,竟然最高点都一样高吗?显然可能性几乎为0。这个如果使用ggplot2的话,可以使用scale='width'强制拉成一样高,但我不推荐,正如我前面说的,不可比较了。默认参数scale='area',积分面积一样,和density curve一样解析,另外的参数scale="count",高度与计数同比例,和histogram一样解析,而scale='width'强制拉成一样高,如果没有在显眼处说明,误导性太强。
画这种图也可以手工拼,这样就简单了。在你需要的情况下,加个灰色背景嘛,最后拼图嘛。当然拼图不一定要在illustrator里拼,比如你用grid,先画好坐标轴,然后水平上定义几个一样大的viewport,每一个violin都画在相应的viewport里面,对于画图函数来说,viewport就是整个画布了(虽然只是画布里的一块区域),所以你要么画violin,要么在画之前先画个矩阵,一路画下来,代码可以直接生成这样的图,但这图每一个violin都是独立画的(当然也不是完全独立,每一个水平上的ylim是有统一的),就算代码一步生成,也跟illustrator里拼没两样。
这里我要教你用ggplot2自动生成,其实解决思路早已推送过,请看《facet_plot:加图层到特定分面,方法二》,也正如我在《什么!你的图上有一双看不见的手》里说的,你们以为我在教ggtree,其实同时在教ggplot2。