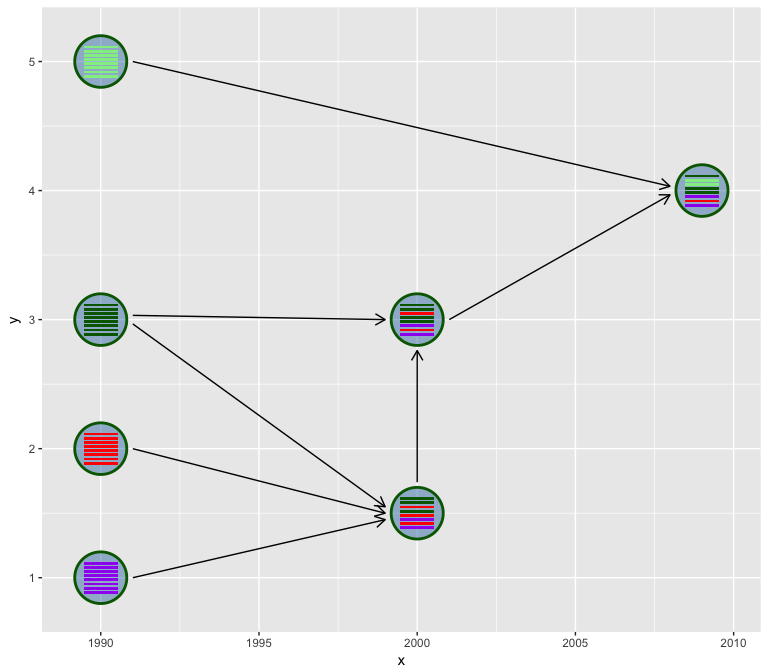
Reassortment is an important strategy for influenza A viruses to introduce a HA subtype that is new to human populations, which creates the possibilities of pandemic.

A diagram showed above (Figure 2 of doi:10.1038/srep25549) is widely
used to illustrate the reassortment events. While such diagrams are
mostly manually draw and edit without software tool to automatically
generate. Here, I implemented the hybrid_plot function for producing
publication quality figure of reassortment events.
library(tibble)
library(ggplot2)
n <- 8
virus_info <- tibble(
id = 1:7,
x = c(rep(1990, 4), rep(2000, 2), 2009),
y = c(1,2,3,5, 1.5, 3, 4),
segment_color = list(
rep('purple', n),
rep('red', n),
rep('darkgreen', n),
rep('lightgreen', n),
c('darkgreen', 'darkgreen', 'red', 'darkgreen', 'red', 'purple', 'red', 'purple'),
c('darkgreen', 'darkgreen', 'red', 'darkgreen', 'darkgreen', 'purple', 'red', 'purple'),
c('darkgreen', 'lightgreen', 'lightgreen', 'darkgreen', 'darkgreen', 'purple', 'red', 'purple'))
)
flow_info <- tibble(from = c(1,2,3,3,4,5,6),
to = c(5,5,5,6,7,6,7))
hybrid_plot(virus_info, flow_info)