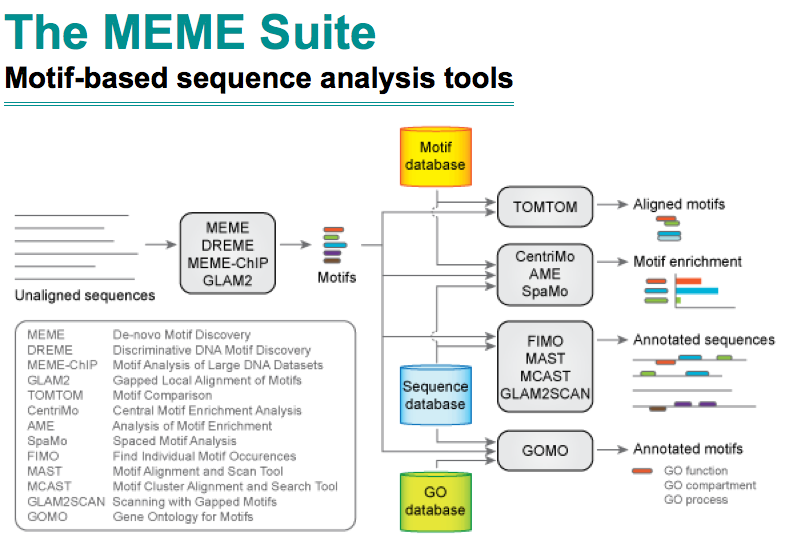
I have played with emoji in R for a while. My solution of using it is different from what implemented in emoGG.
emoGG is a good attemp to add emoji in ggplot2. It render emoji picture (png) and creat a layer, geom_emoji, to add emoji.
In my opinion, emoji should be treated as ordinary font in user interface, albeit it maynot be true internally.
It would be more flexible if we can use emoji as ordinary font and in this way user don’t need to learn extra stuff.