
答案嘛,就是自己画legend啊,你看看legend函数,还不是些简单的代码在画,自己画即可。不要被这些条条框框给套住。
从ggplot2拿legend 为了突破你的想像力,在此处甚至于我要给你个例子,用ggplot2来生成个legend,放在base plot上去。
自己画legend,有何不可?!如果base有现成的函数画图,而你只熟悉ggplot2,你甚至于可以互搞。而互搞,当然要借助于ggplotify包:《ggplotify简史》。
col = colorspace::rainbow_hcl(3) names(col) = unique(iris$Species) library(ggplotify) color = col[iris$Species] p = as.ggplot(~plot(iris$Sepal.Length, iris$Sepal.Width, col=color, pch=15)) library(ggplot2) p2 = ggplot(iris, aes(Sepal.Length, Sepal.Width, color=Species)) + geom_point() + scale_color_manual(values=col, name="") legend = cowplot::get_legend(p2) p + ggimage::geom_subview(x=.7, y=.6, subview=legend) 这里用base plot画了一个图,又用ggplot2画了一个,用cowplot把legend抽出来,然后再用我的另一个包ggimage进行图上嵌图,完事。ggimage实例可以参考这篇《shit,拟合的残差这么大!》。
用ggplot2画legend 上面这例子是不是还不过瘾,因为啊我还是用ggplot2给重新画了一遍,如果你这么看,那么你的脑洞不够大,画是画了,但不一定要画一样的图,比如说有现成的base函数画复杂的图,而你不会用ggplot2画,但不妨碍你画个简单的图,但这简单的图中的图例可以切出来使用啊。
呼应我上面讲到的,图例自己徒手画,我们用ggplot2来来徒手一下。画出下面这个图例:
p2 <- ggplot() + annotate("point", x=1,y=1:3,shape=15, color=col) + annotate("text", x=1.01, y=1:3, label=names(col), hjust=0) + xlim(0.99, 1.2) + ylim(0, 4) + theme_void() 对着代码看,不应该有什么问题,打了三个点,再打了三个相应的文本。然后再让我们把这图当做图例,嵌套到base plot上去。
p + ggimage::geom_subview(x=.
1月
很多人在网络上寻找帮忙而得不到帮助,通常是自己的问题,不要做伸手党,没人欠你一个答案。
2月
我给pheatmap加了个输出为pheatmap对象的功能,再给ggplotify写个方法,让pheatmap的输出转为ggplot,这样就可以不需要grid的知识也可以愉快地进行拼图了。
PS: 之前的版本,也是可以拼的,但你需要一点grid的知识。
很多人用ggpubr,其实应了那一句「越低俗越舒服」的话,无非是因为用起来简单,当然还有另一个因素,是很多公众号搬运了ggpubr的文档,而这些写公众号的,多半也是半吊子水平而已,根本没有分辨的能力。
之前有水生所的小伙伴,在朋友圈发了自己的代码调不好的状态,我评论了一下。

引出了一个text_grob的问题,这就是我说的制造混乱,把gpar几个参数放到gpar外面提供给用户。

我不知道你们看出问题在那里没有,这个text_grob相对于被封装的textGrob没有提供任何额外的功能,而这个封装就等于把gpar的参数给写死了,你想调别的参数,没门,除非你用回textGrob。真是一手好包装。
有人说它有好配色

显然是包装的,特别是说什么杂志的配色。
Thomas Lin Pedersen简直是个天才,最近patchwork动作很大,看到我都准备转投它的怀抱,把旧爱cowplot给扔了。
我们知道patchwork一出来,就推出+号来拼图,最近又搞出了|和/两个操作符。让整个拼图看起来很舒服。
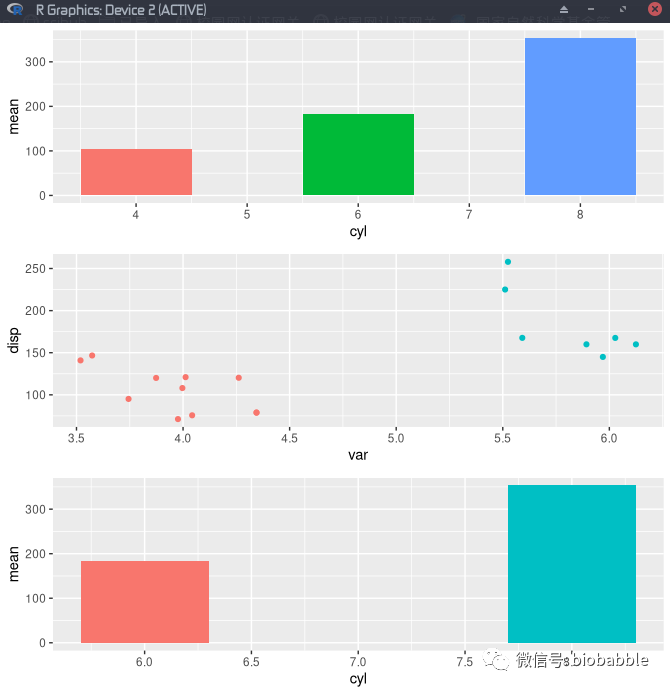
先来画几张图:
library(ggplot2)
p1 <- ggplot(mtcars) +
geom_point(aes(mpg, disp)) +
ggtitle('图一')
p2 <- ggplot(mtcars) +
geom_boxplot(aes(gear, disp, group = gear)) +
ggtitle('图二')
p3 <- ggplot(mtcars) +
geom_point(aes(hp, wt, colour = mpg)) +
ggtitle('图三')
p4 <- ggplot(mtcars) +
geom_bar(aes(gear)) +
facet_wrap(~cyl) +
ggtitle('图四')
ChIPseeker是为ChIP-seq所设计的,因为当年我在做ChIP-seq,一不小心就写了这个包,然而我的知识是有限的,这名字取得太过于限定在ChIP-seq了,其实顺反组的其它类型的测序技术都是支持的,包括DNase-seq和ATAC-seq,此处为了说明我当前的知识是有限的,必须强调不仅限于此,免得出现开头说的这种尴尬。
8月份去开会,有中大肿瘤医院的PI跟我说,他们希望ChIPseeker可以支持ATAC-seq,因为我的ChIPseeker太好用了,然后他们想要用在ATAC-seq上,然而我的包真的是支持的啊。
哈佛大学的网站上有一份ATAC-seq分析指南[1],就明确地写了,ChIPseeker虽然是为ChIP-seq所设计,但对ATAC-seq一样支持得非常好,并且把ChIPseeker列为这份指南的关键步骤之一。
